![]()
![]()
![]()
Virginia Institute of Marine Science, College of William and Mary,
Gloucester Point, Virginia 23062, USA
Background on Haplosporidium nelsoni (MSX) and Perkinsus marinus (Dermo)
Haplosporidium nelsoni
The protozoan oyster pathogen Haplosporidium nelsoni, commonly called MSX, has been responsible for periodic, large-scale oyster mortality in estuaries of the middle Atlantic coast of the United States. The pathogen appeared in Delaware Bay in 1957 and killed 90-95% of all oysters on planted grounds within three years. It appeared in the lower Chesapeake Bay in 1959 and was responsible for the death of about 95% of the oysters in that region by 1962 (Haskin and Andrews, 1988). There have been additional epizootics during periods of extreme drought, especially 1980-81 and 1987-88. The origin of H. nelsoni is uncertain, but there has been considerable speculation that the parasite was introduced with exotic oysters (Andrews, 1984). Although the annual distribution and abundance of H. nelsoni vary with salinity, its general prevalence and intensity have not decreased since the original epizootics and the parasite has now spread along most of the Atlantic coast of the United States.
The widespread distribution and high virulence of H. nelsoni have greatly reduced traditional on-bottom culture and have limited the development of off-bottom oyster aquaculture in high salinity areas. Early diagnosis of H. nelsoni infection is an essential management tool (Ford and Haskin, 1988). Thus, rapid, sensitive diagnostic assays may be critical in avoiding losses to the pathogen. Paraffin-based histological examination, the most commonly used diagnostic technique for H. nelsoni, involves finding parasite cells in one or two 5 µm thick sections through an oyster's visceral mass, gills and mantle. There is a period of several weeks between the time early-summer H. nelsoni infections are heavy enough to be detected histologically and when mortality begins. Therefore, the earlier infections can be detected, the more time is available for oyster growers to decide whether to harvest to limit mortality, or whether to move oysters to low salinity to eliminate the parasite (Ford, 1985). Infections of H. nelsoni acquired in late summer or fall remain nonlethal until spring; early detection would also allow harvest to avoid losses from these late season infections.
Perkinsus marinus
Perkinsus marinus has been a significant cause of mortality of the eastern oyster Crassostrea virginica along the east coast of the United States since the 1950s (Andrews, 1988; Burreson and Ragone Calvo, 1996). The origin of P. marinus is obscure, but it probably has always been an associate of oysters. Along the east coast prior to the late 1980s, P. marinus was restricted to high salinity portions of coastal bays and estuaries south of Delaware Bay, although it apparently was absent from the seaside bays of the eastern shore of Virginia and Maryland (Andrews, 1988). In the Chesapeake Bay, P. marinus was prevalent in the lower bay, but was restricted to the mouths of the major tributaries in Virginia and southern Maryland. There were a few localized concentrations of P. marinus in Maryland, primarily in Fishing Bay and Eastern Bay. The pathogen was observed sporadically in Delaware Bay in the mid-1950s, as a result of importing infected oysters from Chesapeake Bay, but it never caused significant mortality in oysters and appeared to die out as importations stopped in the late 1950s (Ford, 1992). North of Delaware Bay the parasite was absent or at least undetectable. In endemic areas P. marinus has always been responsible for some oyster mortality, but it did not significantly affect harvest most years. The four consecutive drought years in the middle Atlantic region from 1985 through 1988 caused elevated salinities throughout the Chesapeake Bay and allowed P. marinus to spread naturally into areas where oysters were abundant and highly susceptible. Unusually warm winters during the period also contributed to the spread by allowing higher overwintering survival of the pathogen and movement of infected oysters into areas where P. marinus was historically absent also contributed to the problem.
Although drought conditions have abated and rainfall patterns have returned to more or less typical conditions, with wet winters and springs, P. marinus continues to persist tenaciously in most areas of the Chesapeake Bay. Unlike H. nelsoni, which is very susceptible to salinity below 10 ppt, P. marinus can survive in oysters even in areas where salinity drops below 3 ppt for periods of weeks. Thus, even though P. marinus is not causing oyster mortality in many areas, it is present on all beds and if drought conditions return and salinity becomes favorable for development, it will be able to quickly multiply and cause mortality.
Investigators generally rely on Ray's fluid thioglycollate medium (FTM) culture of Perkinsus marinus cells from oyster tissue (rectum, gill, and mantle) for the diagnosis of infected oysters (Ray, 1952). An oyster hemolymph FTM assay was developed to allow monitoring of P. marinus infections without sacrificing the host and to improve quantitation of systemic infections (Gauthier and Fisher, 1990). Using these FTM methods, quantitation relies on accurate counting and the use of a subjective scale developed by Mackin (1962) and modified by Craig et al. (1989). Diagnosis based on FTM methods makes several assumptions. It is assumed that all life stages of P. marinus found in the host are retrieved and that the number of parasites remains constant during incubation in FTM. Furthermore, it is assumed that the distribution of P. marinus in the assayed tissues is representative of the distribution of the parasite throughout the oyster. To overcome this latter assumption, a total body burden assay was developed (Bushek et al., 1994) which employs a procedure using sodium hydroxide to digest oyster tissues after incubation in FTM (Choi et al., 1989). The body burden assay is quantitative and allows enumeration of the total number of parasites in whole oyster homogenates. FTM culture diagnostic methods are relatively simple to perform; however, insensitivity of the assays often causes very light infections to be overlooked.
Molecular diagnostics
There has been increasing interest in developing DNA probes and/or PCR primers as tools for detection of disease agents in aquatic animals. These detection methods have been shown to be more sensitive and specific than previously developed procedures, such as histological examination or immunoassays. DNA probe/PCR technology is ideal for specifically recognizing target DNA regardless of the life history stage present, for identifying cryptic life cycle stages in any potential host organism and for detecting pathogens early in the infection cycle.
The ability to rapidly and accurately detect parasites of oysters has broad implications for both research and industry. As rapid diagnostic methods become more sensitive, early detection of disease agents permits the management of oysters or other cultured species in a manner that is more responsive to patterns of naturally occurring diseases.
However, caution must be exercised in the interpretation of molecular diagnostic results, especially from the polymerase chain reaction (PCR). The extreme sensitivity of this technique may allow amplification of DNA from non-viable or non-pathogenic organisms. A PCR-positive result does not necessarily mean that viable, pathogenic organisms are present or that mortality will eventually occur. More research and experience is needed to fully understand the meaning of PCR-positive results for aquaculture or management. Nonetheless, molecular diagnostic techniques hold great promise for early diagnosis of disease outbreaks in aquaculture and natural populations.
Development of molecular diagnostics for Haplosporidium nelsoni
A sensitive and specific DNA probe and PCR primers have been developed for Haplosporidium nelsoni at the Virginia Institute of Marine Science (Stokes and Burreson, 1995; Stokes et al., 1995).
One difficulty in developing molecular diagnostics for molluscan pathogens in the phylum Haplosporidia is that in vitro culture of these organisms has not been accomplished. Thus it is very difficult to obtain purified organisms for DNA extraction. For our work, plasmodia were concentrated and purified from hemolymph of infected oysters by panning in petri dishes. This technique involves settling of hemocytes, which adhere to the petri dish, allowing the H. nelsoni plasmodia to be decanted. Repeated panning results in an enrichment of plasmodia, although not all hemocytes are excluded. DNA was extracted from the H. nelsoni plasmodia and the small subunit (SSU) rDNA was amplified from the H. nelsoni genomic DNA using the universal primers for eukaryotic 16S-like rDNA.
PCR amplification products were cloned into pCRII and INV?F' using the TA Cloning system (Invitrogen) and subclones were cloned into pUC8 and DH5?. Clones with rDNA inserts were sequenced via the dideoxy chain termination method using the Sequenase kit; the collections of putative H. nelsoni sequence data were assembled into a composite gene sequence using the Gene Jockey software package (GenBank acession number U19538). The variable regions of the SSU rDNA sequence were examined for a section that could be used as a species-specific probe. A putative H. nelsoni-specific oligonucleotide probe, designated MSX1347 (5'-ATGTGTTGGTGACGCTAACCG-3'), was chosen from sequence alignment with the SSU rDNA of Minchinia teredinis, C. virginica, Perkinsus marinus and various protists from GenBank. The probe was synthesized with the incorporation of digoxigenin at the 5' end. In situ hybridizations of this H. nelsoni probe react strongly with H. nelsoni plasmodia (Figure 1) and immature spores, but weakly with mature spores. The probe does not react with oyster tissue, Perkinsus marinus or any of the other haplosporidians present in the middle Atlantic region, including Haplosporidium costale, H. louisiana, or Minchinia teredinis.
Figure 1. In situ hybridization with H. nelsoni DNA probe on histological section of an oyster infected with H. nelsoni.
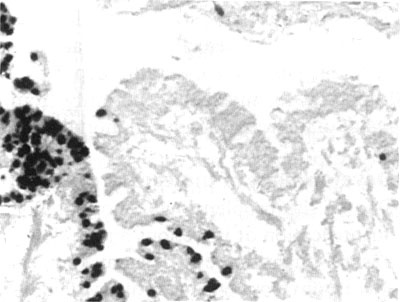
The variable regions of the H. nelsoni SSU rRNA gene were examined for areas which appeared to be species specific and would be appropriate for use as PCR primers. Following identification of suitable priming regions, the putative oligonucleotide sequences were sent as queries to the BLAST electronic mail server ([email protected]) to determine whether the primers would anneal to non-target genes. Two oligonucleotides, designated MSX-A (5'-GCATTAGGTTTCAGACC-3') and MSX-B (5'-ATGTGTTGGTGACGCTA-ACCG-3'), were selected and commercially synthesized. The sequence of the MSX-B primer is the same as the H. nelsoni DNA probe MSX1347 described above. These primers amplify a 564 base pair region of the SSU rRNA gene. Primer sensitivity was tested in ten-fold serial dilutions from 1 ng to 1 fg of cloned H. nelsoni SSU rDNA. A PCR product was easily detectable by agarose gel electrophoresis from a single amplification of 100 fg of cloned H. nelsoni SSU rDNA. Specificity was tested in PCR reactions using genomic DNA from oysters infected and not infected with H. nelsoni, and cloned SSU rDNA of three haplosporidians, M. teredinis, H. costale and H. louisiana. The PCR primers amplified H. nelsoni SSU rDNA from genomic DNA of an infected oyster, but no product was obtained from uninfected oysters or from the other haplosporidians tested.
Validation of PCR diagnosis for Haplosporidium nelsoni
During May 1995, 400 oysters uninfected with H. nelsoni were imported to the lower York River estuary, Virginia. These oysters were examined monthly through December for H. nelsoni using three diagnostic techniques-paraffin histology, the routine diagnostic technique (i.e. gold standard), PCR of oyster hemolymph and PCR of gill and mantle tissue. A sample of 25 oysters was collected each month. The oyster shells were notched and 0.5 ml of hemolymph was withdrawn from the adductor muscle sinus with a needle and syringe for DNA extraction. Each oyster was then shucked and a 0.25 g piece of gill and mantle tissue was removed for DNA extraction. The remaining oyster was preserved in Davidson's AFA fixative for histological analysis. Results of the analyses are shown in Figure 2. PCR of hemolymph detected H. nelsoni infections two months earlier (May) than either PCR of tissue or histology (July). PCR of both hemolymph and tissue always detected a higher prevalence of infection that histology, although during November and December, when all infections are typically systemic, there was little difference in prevalence among the three techniques. Overall, infections were detected in 129 oysters by one of the three methods. Fifty five infections were detected by one of the two PCR techniques that were negative by histological examination and two infections were detected by histology that were negative by PCR.
These results demonstrate that PCR diagnosis is much more sensitive than histology and allowed infections to be detected much earlier in the natural infection cycle (For another interpretation from a different perspective see the paper by Dan Fegan in this report). This early detection can be very important for mitigating mortality from the disease. The decline in prevalence during August is the result of H. nelsoni-induced mortality that typically occurs during late July as infections intensify.
Development of molecular diagnostics for Perkinsus marinus
One of the problems with normal PCR is that it is not quantitative and there is much interest in quantification of infections and in detection and quantification of P. marinus in natural water samples. A semi-quantitative PCR assay for P. marinus was developed by Marsh et al. (1995), so our interest was in developing a quantitative analysis (Yarnall et al., 1999) called quantitative competitive PCR (QCPCR).
Figure 2. Prevalence of
Haplospordium nelsoni in oysters through a natural infection cycle in
Chesaspeake Bay, Virginia as determined by three different diagnostic
techniques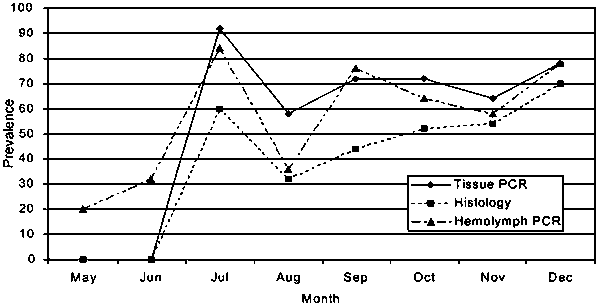
Limitations of precise quantitation using PCR techniques stem from the lack of consistent initial exponential increase in product during the amplification reaction. Replicate samples can be subjected to variations in reaction conditions such as inhibitor concentrations, polymerase integrity, and even thermal cycler block positions. These differences can result in variation in amplification efficiencies that may obscure differences in the amounts of DNA or RNA that are being measured and thus preclude accurate quantitation. In competitive PCR, the standard and target DNA are present in the same reaction tube. The target DNA and internal standard DNA compete for the same DNA polymerase and deoxynucleotide triphosphates. In addition, the two DNA templates are equally affected by tube to tube variations in the PCR conditions including inhibitor effects. Furthermore, competitive PCR overcomes the need to perform quantitation in the exponential phase since both the standard and the target are equally affected by the changes in amplification parameters that occur as the reactions enter the plateau phase. Temperature cycling into the plateau phase, therefore, does not interfere with quantitation and even increases the sensitivity of the assay.
Ultimately, quantitation by competitive PCR involves a set of reactions that include a constant volume aliquot of unknown DNA concentration and a dilution series of known concentrations of the standard competitor DNA. At the point where the molar amounts of the two products are equivalent, the amount of original target DNA present in the sample is equivalent to the amount of standard initially added. Thus, quantitation of the unknown DNA is based on the attainment of an equivalence point at a known concentration of standard competitor DNA. Internal competitive standards for quantitative PCR typically have sequences that are homologous to the target nucleic acids and amplify with the same or slightly modified primers. The target and the competitor, therefore, amplify with the same kinetics. Altering the size of the competitor molecule relative to the target sequence has been shown to be an excellent method for competitor. This method eliminates the need to use restriction enzymes with variable digestion efficiencies to cut competitor molecules engineered with unique sites. Using automated sequencers, a one base pair size difference between PCR products can be detected, allowing extremely similar competitor molecules to be utilized.
Primers were derived from the published P. marinus DNA sequences for the ribosomal RNA gene (Fong et al., 1993) and the adjacent internal transcribed spacer (ITS-1) region (Goggin, 1994). The primers, designated PER-18S (5'CCTA-CGGGATTGGTTGTATCAG3') and PER-ITS (5'CATCTCGCAACTCTCTAACA-AAAG3') specifically amplified a 1210 base pair fragment of DNA from within the SSU rRNA gene to within the ITS-1 of the ribosomal DNA region. The primers were tested for sensitivity with cloned SSU rDNA derived from pure cultures of P. marinus. As little as 0.005 fg of P. marinus DNA was detected. They were tested for specificity with DNA from cultured cells of P. atlanticus, from cultured cells of a variety of P. marinus isolates from along the east coast of the United States, and from a variety of cultured dinoflagellates. The primers amplified DNA from all isolates of P. marinus, but not from P. atlanticus or any of the dinoflagellates.
A competitor DNA molecule was constructed using the PCR primers PER-18S and ITS-MUT (5'CATCTCGCAACTCTCTAACAAAAGagcaagagagagcGAGACCGC-TG3'). The latter (ITS-MUT) contained the entire sequence of the PER-ITS primer; however, the ITS-MUT primer created a gap of thirteen nucleotides in the sequence to be amplified by linking to a region of sequence several bases upstream, within the ITS-1 region. The PER-18S and ITS-MUT PCR product was and cloned.
The QCPCR assay involved a two-phase approach. Titration spanning a broad range of competitor dilutions was performed to obtain a rough estimate of the amount of DNA and then a second titration over a narrower range was performed for precise quantitation. A standard curve was prepared by extracting DNA from a quantified number of cultured P. marinus cells (102 to 106 cells). The standard curve related the number of cells to the amount of DNA as determined by QCPCR.
Validation of PCR diagnostics for Perkinsus marinus
Target DNA was obtained from oysters collected from the lower Chesapeake Bay, Virginia. Oyster shells were notched and hemolymph samples were withdrawn with needle and syringe. Approximately 0.6 ml of hemolymph was divided equally for DNA extraction and for hemolymph Fluid Thioglycollate (FTM) analysis. Oysters were then shucked and 0.25 g of gill/mantle tissue was removed for DNA extraction and an equal amount for standard FTM analysis. The remaining oyster tissue was processed for the total body burden FTM analysis (Fisher and Oliver, 1996). Results are shown in Table 1. For the QCPCR results, any number >0 but <1 was assigned a value of 1 cell.
Ray's FTM tissue assay diagnosed only 19 infections while the body burden whole oyster tissue assay diagnosed infections in 24 of the 25 oysters. The FTM hemolymph assay detected 22 infections. QCPCR with both the gill and mantle tissue and the hemolymph DNA detected 24 infections. The Total Body Burden analysis (the gold standard) detected infections in 24 of the 25 oysters. The FTM tissue assay, the assay used routinely by most laboratories, detected infections in only 19 of the oysters, while FTM of oyster hemolymph detected infections in 22 of the oysters. QCPCR of both tissue and hemolymph detected infections in 24 of the oysters, equal to the Total Body Burden analysis. Quantitation of QCPCR compared to Total Body Burden or FTM hemolymph analysis was generally good. Values were generally within one order of magnitude and were often the same order of magnitude. Considering that infections ranged over 7 orders of magnitude, the results were generally comparable.
PCR diagnosis was not more sensitive than the total body burden assay for P. marinus, although it was equally sensitive. This is undoubtedly because the total body burden assay is an analysis of the number of P. marinus cells in the entire oyster whereas for PCR only a small piece of tissue or a small aliquot of hemolymph was used for analysis. Thus, very light or localized infections may be missed in PCR. Therefore it is perhaps surprising that PCR analysis of both tissue and hemolymph were as sensitive as the body burden assay.
References
Andrews, J.D. (1984). Epizootiology of diseases of oysters (Crassostrea virginica), and parasites of associated organisms in eastern North America. Helgoländer Meeresuntersuchungen 37, 149-166.
Andrews, J.D. (1988). Epizootiology of the disease caused by the oyster pathogen Perkinsus marinus and its effects on the oyster industry. American Fisheries Special Publication 18, 47-63.
Burreson, E.M. and Ragone Calvo, L.M. (1996). Epizootiology of Perkinsus marinus disease of oysters in Chesapeake Bay with emphasis on data since 1985. Journal of Shellfish Research 15, 17-34.
Bushek, D., Ford, S.E. and Allen, S.K. (1994). Evaluation of methods using Ray's fluid thioglycollate medium for diagnosis of Perkinsus marinus infection in the eastern oyster, Crassostrea virginica. Annual Review of Fish Diseases 4, 201-217.
Choi, K.S., Wilson, E.A., Lewis, D.H., Powell, E.N. and Ray, S.M. (1989). The energetic cost of Perkinsus marinus parasitism in oysters: quantification of the thioglycollate method. Journal of Shellfish Research 8, 125-131.
Fisher, W.S. and Oliver, L.M. (1996). A whole-oyster procedure for diagnosis of Perkinsus marinus disease using Ray's fluid thioglycollate culture medium. Journal of Shellfish Research 15, 109-117.
Fong, D., Rodrigues, R., Koo, K., Sun, J., Sogin, M.L., Bushek, D., Littlewood, D.T.J. and Ford, S.E. (1993). Small subunit ribosomal RNA gene sequence of the oyster parasite Perkinsus marinus. Molecular Marine Biology and Biotechnology 2, 346-350.
Ford, S.E. (1985). Effects of salinity on survival of the MSX parasite Haplosporidium nelsoni (Haskin, Stauber and Mackin) in oysters. Journal of Shellfish Research 5, 85-90.
Ford, S.E. (1992). Avoiding the transmission of disease in commercial culture of molluscs, with special reference to Perkinsus marinus (Dermo) and Haplosporidium nelsoni (MSX). Journal of Shellfish Research 11, 539-546.
Ford, S.E. and Haskin, H.H. (1988). Management strategies for MSX (Haplosporidium nelsoni) disease in eastern oysters. In Disease Processes in Marine Bivalve Molluscs. W. S. Fisher (ed.). American Fisheries Society Special Publication 18, 249-256.
Gauthier, J.D. and Fisher, W.S. (1990). Hemolymph assay for diagnosis of Perkinsus marinus in oysters Crassostrea virginica. Journal of Shellfish Research 9, 367-372.
Goggin, C.L. (1994). Variation in the two internal transcribed spacers and 5.8 S ribosomal RNA from five isolates of the marine parasite Perkinsus (Protista, Apicomplexa). Molecular and Biochemical Parasitology 65, 179-182.
Haskin, H.H. and Andrews, J.D. (1988). Uncertainties and speculations about the life cycle of the eastern oyster pathogen Haplosporidium nelsoni (MSX). In: Fisher, W. S. (ed.), Disease Processes in Marine Bivalve Molluscs. American Fisheries Society Special Publication 18, 5-22.
Mackin, J.G. (1962). Oyster disease caused by Dermocystidium marinum and other microorganisms in Louisiana. Publication of the Institute of Marine Science, University of Texas 7, 132-229.
Marsh, A.G., Gauthier, J.D. and Vasta, G.R. (1995). A semiquantitative PCR assay for assessing Perkinsus marinus infections in the eastern oyster, Crassostrea virginica. Journal of Parasitology 81, 577-583.
Ray, S.M. (1952). A culture technique for the diagnosis of infection with Dermocystidium marinum (Mackin, Owen, and Collier) in oysters. Science 114, 360-361.
Stokes, N.A. and Burreson, E.M. (1995). A sensitive and specific DNA probe for the oyster pathogen Haplosporidium nelsoni. Journal of Eukaryote Microbiology 42, 350-357.
Stokes, N.A., Siddall, M.E. and Burreson. E.M. (1995). Detection of Haplosporidium nelsoni (Haplosporidia: Haplosporidiidae) in oysters by PCR amplification. Diseases of Aquatic Organisms 23, 145-152.
Yarnall, H.A., Reece, K.S., Stokes N.A. and Burreson. E.M. (1999). A quantitative competitive polymerase chain reaction assay for the oyster pathogen Perkinsus marinus. Journal of Parasitology (submitted).
Table 1. Diagnosis of Perkinsus marinus infection by five different techniques.
|
Oyster # |
FTM tissue |
Total body |
QCPCR gill |
FTM |
QCPCR |
|
1 |
1.0 |
3332 |
3090 |
27 |
2 |
|
2 |
5.0 |
11,379,338 |
4,466,840 |
3689 |
920 |
|
3 |
0.5 |
76 |
468 |
0 |
1 |
|
4 |
0.5 |
3397 |
30 |
13 |
1 |
|
5 |
0.5 |
2040 |
275 |
7 |
1 |
|
6 |
4.0 |
595,104 |
22,909 |
4622 |
224 |
|
7 |
1.0 |
102,237 |
2042 |
7 |
22 |
|
8 |
1.0 |
5437 |
589 |
30 |
1 |
|
9 |
5.0 |
1,235,772 |
60,256 |
62,833 |
1125 |
|
10 |
1.0 |
427,083 |
37,154 |
7200 |
398 |
|
11 |
5.0 |
4,217,826 |
128,825 |
17,778 |
2618 |
|
12 |
3.0 |
663,727 |
134,896 |
2200 |
867 |
|
13 |
5.0 |
38,725,490 |
2,187,760 |
5,866,667 |
20,512 |
|
14 |
0.0 |
44,538 |
324 |
40 |
1 |
|
15 |
2.0 |
274,296 |
9333 |
7222 |
58 |
|
16 |
4.0 |
258,763 |
15,136 |
84,444 |
12,503 |
|
17 |
1.0 |
3219 |
427 |
37 |
25 |
|
18 |
0.5 |
120,622 |
158 |
27 |
15 |
|
19 |
1.0 |
0 |
* |
0 |
0 |
|
20 |
0.5 |
185 |
8 |
7 |
1 |
|
21 |
0.0 |
640 |
49 |
7 |
1 |
|
22 |
0.0 |
1087 |
155 |
13 |
1 |
|
23 |
0.0 |
4167 |
7 |
0 |
1 |
|
24 |
0.0 |
18 |
0 |
3 |
1 |
|
25 |
0.0 |
14 |
* |
40 |
* |
* Denotes a positive result by QCPCR but the below the level of quantitation (<0.005 fg).
† The Mackin scale (0-5) is a subjective rank based on the number of P. marinus cells observed with a microscope over all fields examined; the higher the Mackin rank, the heavier the infection.
![]()
![]()
![]()