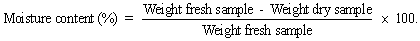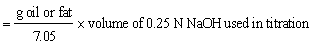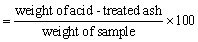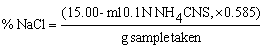![]()
![]()
![]()
1. INTRODUCTION
2. QUALITY CONTROL PROCEDURES
3. RAW MATERIAL AND FINISHED PRODUCT STORAGE
4. METHODS OF ANALYSIS
5. REFERENCES
K. W. Chow
Food and Agriculture Organization
Rome, Italy
Quality control in the compound feed industry not only involves the verification of quality standards established for each feed ingredient as it is received into storage in the mill, but also involves the close monitoring of the quality of ingredients through the period of storage prior to usage and during its processing. Quality control continues as ingredients are merged during the mixing process and as they finally go into storage as compound feed.
Feed manufacturing enterprises in developing countries operate under more difficult conditions than in developed countries. Problems of finance, management, and technical expertise aside, great difficulties in obtaining adequate supplies of raw materials are frequently encountered. Choices may also be restricted largely to feeds that are produced locally, since governments are reluctant to divert scarce foreign exchange for imports. Prices and availability of domestically produced feedstuffs are often subject to seasonal fluctuations while quality may also vary widely. Needed imports are made through agents with little experience in or responsibility for the quality of materials actually shipped. As a result, the need for quality control of both raw materials and finished products is greater for feed manufacturers in developing countries.
The purpose of quality control of raw materials is to ensure that minimum contract specifications are met. More precisely, it provides knowledge concerning the exact composition of raw materials and the level's of toxic substances normally present so that mixed feeds of the required nutritive value can be safely processed from them. These specifications are usually determined by a team consisting of the nutritionist, management personnel, and the quality control manager. The specifications relate to nutrient quality, cost, and the quality desired in the feedstuffs. Once the specifications are decided upon, they must be used and adhered to. Such a decision must be based on realistic reasoning, since it is useless to have specifications which are so narrow as to make purchasing impossible, or are so broad as to present severe problems for diet formulation.
Most contemporary feeds for livestock are formulated with the aid of a computer which calculates the cheapest product obtainable from available raw materials within constraints laid down for the nutritive value, toxicity, and palatibility of the final product (see chapters on Feed Formulation). Such least cost formulations are possible only when the composition of each raw material is known with a high degree of precision and when good quality control is maintained.
2.1.1 Preliminary inspection
Materials received at the mill should be subjected to a thorough physical inspection to determine the following:
(a) evidence of wetting - mould growth confirms water damage;
(b) presence of scrap metals, stones, dirt, or other non-biological contaminants; and
(c) presence of insects.
Moisture content of feed grains should be determined by one of the rapid procedures commercially available. Any consignment exceeding 13 percent moisture is especially prone to insect and mould attack and should be separated from other stored ingredients. Preferably, materials with high moisture content should not be taken into the store until after drying.
2.1.2 Sampling
Sampling is the most important activity in quality control, because no analysis can be better than the sample from which it is made. Hence, proper procedures for taking representative samples for quality evaluation are essential.
Sampling of bagged ingredients is done with a spear probe. The probe is inserted diagonally and as horizontally as possible, from one corner of the bag to the other. In lots of 1-10 bags, all bags are sampled. In larger lots, 10 percent of all bags are sampled. Materials received in bulk are sampled by using a scoop, according to the size of the consignment. For smaller than 10 t consignments, two samples per ton are taken. Larger consignments, up to 100 t, require one sample per ton or one sample for every two tons depending on the size of the consignment.
Samples taken in the above manner should be pooled, thoroughly mixed, and then reduced in size by quartering to between 1 and 2 kg in weight.
Oil cakes and other coarse materials are sampled by random selection of pieces from different parts of the entire consignment. Five pieces per ton of materials are considered sufficient. The pieces should then be ground, mixed thoroughly, and the sample reduced in size to between 1 and 2 kg - as outlined above.
Samples submitted to the quality control laboratory should be placed in tightly sealed containers. Prior to chemical analysis, the samples must be reduced to a powder using a Waring type blender or a mortar.
2.1.3 Tests required
For feedstuffs likely to contribute to both the protein and energy content of the final product, a "Weende" proximate analysis is usually conducted. This determines the moisture, crude protein, lipid, crude fibre, ash, and nitrogen-free extract content of the feedstuff. Methods of analysis are given in Section 4.
Additional tests should be carried out on materials having a high ash content to determine the proportion of acid insoluble ash present, thus providing an indication of the amount of sand or other dirt present. This will also enable detection of any deliberate contamination of expensive feedstuffs such as fishmeal. Salt (NaCl) analysis of fish meal is also required for these same reasons, as well as to avoid excessive levels of sodium in the final diet.
Calcium and phosphorus determinations are routinely conducted on mineral feeds, such as bone meal, calcium phosphate and calcium carbonate sources. Other feedstuffs such as fish meal, and meat and bone meal are also analyzed for these two mineral elements.
2.1.4 Other tests
The protein requirements of some cultured animal species are more exacting than others. For example, fish not only require higher protein levels in their diets but the protein quality requirements are also higher. The determination of amino acid content is a lengthy and complicated process requiring highly specialized, and expensive, equipment. However, it is advisable to test feedstuffs, such as fish meal, for lysine availability.
Certain feedstuffs contain natural toxins that, at high enough levels, are growth inhibitory and sometimes fatal to the animal consuming them. Principal among these are:
(a) ureaseAn enzyme found in raw soybeans which produces toxicity through the hydrolysis of urea to toxic ammonia. The toasting process in soybean meal manufacture destroys the enzyme.(b) gossypol
An endogenous toxin present in the gland of cottonseed which persists during production of the meal unless removed by a special process, or, unless the cottonseed is a glandless variety.(c) isothiocyanates
cyanogenic glycosides are found in linseed and cassava. Much of the toxicity is eliminated during processing of the raw material.(d) aflatoxin
Aflatoxin is a class of extremely potent toxins produced by the mould Aspergillus flavus. Aflatoxin may be present in any materials produced and stored under hot and humid conditions and is usually found in groundnut cake, palm kernel cake, copra cake, and maize (which have not been properly dried after harvest).
Molasses is a common feedstuff in the tropics. Periodic analysis is necessary to establish its sugar content. Molasses sometimes contain an undesirably high level of potassium and occasional checks on this should be conducted.
Fats are rapidly oxidized in the warm tropics. Rancidity causes off-flavour in feeds. Levels of free fatty acids in expeller cakes, such as palm oil and copra cakes, should be frequently checked. Unrefined palm oil or palm oil sludge, frequently used to increase the energy content of compounded feeds, contain high levels of free fatty acids.
Trace mineral and vitamin supplements form an important part of balanced diets for both fish and livestock. The availability of these feed components from reliable sources makes it quite unnecessary to perform routine checks on their quality unless large volume purchases are involved. Sending samples to a reliable commercial laboratory obviates the great expenses of equipping a laboratory for such testing.
Quality control of the finished products consists of determining the manufacturing process to ensure that ingredients were added in the proportions required by the formulation. Inhomogeneity of the final product, due to improper mixing or unwanted ingredient separation, thus, can be detected. If it can be established that raw materials conform with specifications and that process control is adequate, then only periodic checks on the finished products are necessary.
2.2.1 Preliminary inspection
Most modern mills are equipped with sieves and magnets along the material flow lines for removal of tramp metal, rocks, and other scrap contaminants. However, smaller operations may not have such features, and physical inspection of the finished product should be carried out to determine the presence of such contaminants. Any detection of foreign contaminants should be brought to the attention of the mill supervisor who could then determine if the contaminants originated in the raw material or if they were the result of improper maintenance within the mill premises.
2.2.2 Sampling
To detect product inhomogeneity and significant ingredient separation during the manufacturing process, sampling should be obtained during bagging-off time by taking a handful from every fifth bag of 40-50 bags and pooling the individual samplings. Testing for variability is best conducted by probing the bottom, middle, and top of a bag with a short probe. Tests may be made on each sample, or the samples from the same bags may be combined and mixed before analysis depending upon the question to be answered.
The size of finished feed samples need only be half that required for raw materials, i.e., 0.5-1.0 kg after quartering.
Samples submitted for chemical analysis should be placed in tightly sealed containers.
2.2.3 Tests required
It is not uncommon for feed to be despatched to the customer on the same day it is manufactured, a day or two before chemical analysis on the product is completed. Therefore, one senses the importance of proper raw material quality control and the apparent irrelevance of quality testing on finished products. Nonetheless, quality control of finished products is necessary because it serves two important functions:
(a) it checks the manufacturing process, and
(b) it checks the quality specifications, or claims, established for the finished products.
If raw material quality control is properly conducted and if process control is adequate, then the only chemical tests required of finished products on a regular basis are for moisture and crude protein. Periodic inspections on other components of the "Weende" proximate analysis should also be scheduled.
Both raw materials and finished feeds undergo deteriorative changes during storage. For raw materials, these changes represent a direct economic loss because of the resultant decrease in nutritive value. Finished products undergo changes which not only lower their nutritive value below minimum specifications but also affect their palatability and appearance.
Due to seasonal fluctuations in availability and price, it is often necessary to maintain large inventories of raw materials if demands are to be continuously satisfied at stable price levels. Six-month inventories of stocks of imported raw materials are not uncommon. In the tropics, such lengthy storage periods present special problems, because the two environmental factors which most affect deteriorative changes in stored feeds are temperature and relative humidity. The rates of chemical changes that normally occur are increased with increasing ambient temperatures. Insect and mould growth are also favoured by the warmer and more humid I climate of the tropics. In addition, there is a distinct relationship between insect activity and chemical changes in stored feedstuffs. A more thorough discussion of deteriorative changes in stored feed materials is presented in Chapter 13. It suffices to state here that to minimize deterioration of stored raw materials and finished feeds, it is essential to keep them as cool and dry as possible) to minimize infestation by insect and rodent pests. Accordingly, warehouses should be constructed in such a way that the interior can be kept cool and dry with adequate ventilation to reduce micro-climatic variations created by the presence of the stored materials.
4.1 Moisture
4.2 Crude Protein (Kjeldahl Method)
4.3 Crude Fat
4.4 Crude Fibre
4.5 Ash
4.6 Sodium Chloride
4.7 Molasses Analysis
4.8 Anti-metabolite and Toxins in Feeds
Weigh and place 4-5 g of the sample in a covered, flat, aluminium dish. Dry to constant weight at 100-105°C in a drying oven.
Reagents:
(a) sulphuric acid (98%), nitrogen free,(b) potassium sulphate, reagent grade,
(c) mercuric oxide, reagent grade,
(d) paraffin wax,
(e) sodium hydroxide, 40% solution,
(f) sodium sulphide, 4% solution,
(g) pumice chips,
(h) boric acid/indicator solution. Add 5 ml of indicator solution (0.1% methyl red and 0.2% bromocresol green in alcohol) to 1 litre saturated boric acid solution, and
(i) hydrochloric acid standard solution (0.1N).
Apparatus:
(a) macro Kjeldahl digestion and distillation units,
(b) Kjeldahl flasks (500 ml capacity or larger), and
(c) conical flasks, 250 ml.
Method:
Accurately weigh 1 g of sample into a digestion- flask. Add 10 g potassium sulphate, 0.7 g mercuric oxide (pre-measured catalyst tablets containing these two reagents are available), and 20 ml sulphuric acid. Heat the flask gently at an inclined angle until frothing subsides and then boil until the solution clears. Continue boiling for an additional half hour. If the frothing is excessive, a small amount of paraffin wax may be added.
On cooling, add about 90 ml distilled water, recool, add 25 ml sulphide solution, and mix. Add a small piece of boiling chip to prevent bumping and 80 ml of sodium hydroxide solution while tilting the flask so that two layers are formed. Connect rapidly to the condenser unit, heat, and collect distilled ammonia in 50 ml boric acid/indicator solution. Collect 50 ml of distillate. On completion of distillation, remove the receiver (wash condenser tip) and titrate against standard acid solution.
Calculation:
Nitrogen content of sample (%)

Reagents and equipment:
(a) petroleum ether (b.p. 40-60°C),
(b) extraction thimbles, and
(c) Soxhlet extraction apparatus.
Method:
Weigh into an extraction thimble 2-3 g of the dried sample (residue from dry matter determination can be used). Place the thimble inside the Soxhlet apparatus. Connect a dry pre-weighed solvent flask beneath the apparatus and add the required quantity of solvent and connect to condenser. Adjust the heating rate to give a condensation rate of 2 to 3 drops/s and extract for 16 h. (The extraction time may be reduced to a minimum of six h by increasing the condensation rate.) On completion, remove the thimble and reclaim ether using the apparatus. Complete the removal of ether on a boiling bath and dry flask at 105°C for 30 min. Cool in a desiccator and weigh.
Calculation:
Crude fat (% of DM)
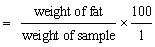
4.3.1 Free fatty acids
Reagents and apparatus:
(a) ethyl alcohol,
(b) phenolphthalein (1% soln, in alcohol),
(c) sodium hydroxide (0.25N), and
(d) stoppered flasks, 250 ml. Method:
Weigh oil or fat into a stoppered flask and add 50 ml alcohol previously neutralised by adding sufficient 0.25N sodium hydroxide to give a faint pinkish colour with phenolphthalein (2 ml). Titrate with sodium hydroxide and vigorous shaking until a permanent faint pink colour appears.
Calculation:
Free fatty acids % (as oleic acid)
Retain extracted sample for crude fibre analysis and extracted fat for free fatty acid determinations.
Reagents:
(a) sulphuric acid solution (0.25 N),
(b) sodium hydroxide solution (0.313N),
(c) antifoam reagent (Octyl alcohol),
(d) ethyl alcohol, and
(e) hydrochloric acid, 1% v/v.
Apparatus:
(a) beakers, 600 ml tall-sided,
(b) round-bottom flask condenser unit,
(c) Buchner flasks, 1 litre,
(d) Buchner funnels. Hartley 3 section pattern,
(e) crucibles, silica with porous base, and
(f) rubber cones to fit above.
Method:
Weigh about 2 g of the dried, fat-free sample into a 600 ml beaker. Add 200 ml of hot sulphuric acid, place the beaker under the condenser, and bring to, boiling within 1 min. Boil gently for exactly 30 min, using distilled water to maintain volume and to wash down particles adhering to the sides. Use antifoam if necessary. Filter through Whatman No. 541 paper in a Buchner funnel, using suction, and wash well with boiling water. Transfer residue back to beaker and add 200 ml hot sodium hydroxide solution. Replace under the condenser and again bring to boil within 1 min. After boiling for exactly 30 min, filter through porous crucible and wash with boiling water, 1% hydrochloric acid and then again with boiling water. Wash twice with alcohol, dry overnight at 100°C, cool, and weigh. Ash at 500°C for 3 h, cool, and weigh. Calculate the weight of fibre by difference.
Calculation:
Crude fibre (% of fat-free DM)
Weigh a 2 g sample into a dry, tared porcelain dish and then place in a muffle furnace at 600°C for 6 h. Cool in a desiccator and weigh.
Calculation:
Ash (%)
Acid soluble and insoluble ash Reagents and apparatus:
(a) hydrochloric acid (1-2.5 v/v),
(b) filter paper, ashless, and
(c) dishes, porcelain.
Method:
Use the residue obtained from the ash determination. Boil with 25 ml hydrochloric acid, taking care to avoid spattering, filter through ashless filter paper, and wash with hot water until acid free. Place filter paper and residue into a dry, tared porcelain dish and place in a muffle furnace at 600°C for 2 h or until carbon free.
Calculation:
Acid insoluble ash (%)
4.5.1 Calcium Reagents:
(a) hydrochloric acid (1-3 v/v),
(b) nitric acid (70%),
(c) ammonium hydroxide (1-1 v/v),
(d) methyl red indicator (dissolve 1 g in 200 ml alcohol),
(e) ammonium oxalate (4.2% solution),
(f) sulphuric acid (98%), and
(g) standard potassium permanganate solution (0.05 N).
Apparatus:
(a) porcelain dishes,
(b) volumetric flasks, 250 ml,
(c) beakers, 250 ml,
(d) quantitative filter paper and funnels, and
(e) burette.
Method:
Weigh 2.5 g of finely ground material into a porcelain dish and ash as above (alternatively use residue from ash determination). Add 40 ml hydrochloric acid and a few drops of nitric acid to the residue, boil, cool, and transfer to a 250 ml volumetric flask. Dilute to volume and mix.
Pipette a suitable aliquot of the solution (100 ml for cereal feeds, 25 ml for mineral feeds) into a beaker, dilute to 100 ml and add 2 drops of methyl red. Add ammonium hydroxide one drop at a time until a brownish orange colour is obtained, then add two drops of hydrochloric acid to give a pink colour. Dilute with 50 ml water, boil, and add while stirring 10 ml of hot 4.2 percent ammonium oxalate solution. Adjust the pH with acid to bring back pink colour if necessary. Allow precipitate to settle out, and filter, washing precipitate with ammonium hydroxide solution (1.50 v/v). Place the filter paper with precipitate back in beaker and add a mixture of 125 ml water and 5 ml sulphuric acid. Heat to 70°C and titrate against the standard permanganate solution.
Calculation:
Calcium (%)
4.5.2 Phosphorous Reagents:
(a) Molybdovanadate reagentDissolve 40 g ammonium molybdate 4H2O in 400 ml hot water and cool. Dissolve 2 g ammonium metavanadate in 250 ml hot water, cool, and add 450 ml 70 percent perchloric acid. Gradually add the molybdate solution to the vanadate solution with stirring and dilute to 2 litres.(b) Phosphorous standards
Prepare stock solution by dissolving 8.788 g potassium dihydrogen orthophosphate in water and making up to 1 litre. Prepare the working solution by diluting the stock 1 in 20 (working concentrate 0.1 mg P/ml).
Apparatus:
(a) spectrophotometer to read at 400 mm, and
(b) graduated flasks, 100 ml.
Method:
Pipette an aliquot of the sample solution prepared as for the calcium determination into a 100 ml flask and add 20 ml of the molybdovanadate reagent. Make up the volume, mix, and let stand for 10 min. Transfer aliquote of the working standard containing 0.5, 0.8, 1.0, and 1.5 mg phosphorus to 100 ml flasks and treat as above. Read sample at 400 my setting the 0.5 mg standard at 100 percent transmission. Determine mg phosphorus in each sample aliquot from a standard curve.
Reagents:
(a) standard 0.1 N silver nitrate solution,
(b) standard 0.1 N ammonium thiocyanate solution,
(c) ferric indicator - saturated aqueous solution of ferric aluminium,
(d) potassium permanganate solution - 6% w/v,
(e) urea solution - 5% w/v, and
(f) acetone (A.R. grade).
Method:
Weigh 2 g sample into a 250 ml conical flask. Moisten sample with 20 ml water and then add, by pipette, 15 ml 0.1 N silver nitrate solution and mix well. Add 20 ml concentrated nitric acid and 10 ml potassium permanganate solution and mix. Heat mixture continuously until liquid clears and nitrous fumes are evolved; then cool. Add 10 ml urea solution and allow to stand for 10 min. Add 10 ml acetone and 5 ml ferric indicator, and back titrate the excess silver nitrate with the 0.1 N thiocyanate solution to the red brown end point.
Calculation:
Calculate results as sodium chloride,
4.7.1 Total sugars
Reagents
(a) Fehling's solution (Soxhlet modification)(i) Dissolve 34 639 g of copper sulphate 5 H2O in water and make up to 500 ml. Filter, and(ii) Dissolve 173 g of Potassium sodium tartrate 4 H2O and 50 g sodium hydroxide in water, dilute to 500 ml, stand for two days, and filter through prepared asbestos.
(b) Invert sugar standards
Prepare stock solution by adding 5 ml of hydrochloric acid (sp.g 1.18) to 9.5 g of sucrose in solution and dilute to about 100 ml. After storing for two days at room temperature, dilute to 1 litre. Prepare working solutions (5 mg/ml) by pipetting 100 ml of the stock solution into a 200 ml volumetric flask, and neutralising with 20 percent sodium hydroxide using phenolphthalein as the indicator. Dilute to mark, and mix.(c) hydrochloric acid (sp. g 1.18),
(d) hydrochloric acid (0.5 N),
(e) sodium hydroxide (20%),
(f) phenolphthalein indicator (1% solution in alcohol), and
(g) methylene blue indicator (1% aqueous solution).
Apparatus:
(a) electric heater, and
(b) conical flasks, 300 ml.
Method:
Dissolve 8 g of liquid molasses and make up to 500 ml. Carry out an acid hydrolysis on 100 ml of the filtrate by adding 5 ml of hydrochloric acid (sp. g 1.18) and allow to stand for 24 h. Neutralise with sodium hydroxide (20 percent) using phenolphthalein as indicator, and then dilute to 200 ml.
Standardisation of Soxhlet solution. Pipette 10 ml of Soxhlet solutions (a) and (b) into a conical flask, mix, and add 30 ml of water. Add from a burette a volume of working standard that is almost sufficient to reduce the copper in the Soxhlet solution. Bring to boiling and continue boiling for two minutes. Add four drops of methylene blue and rapidly complete the titration, while still boiling, until a bright orange colour is resumed. Repeat several times and determine the volume of solution required to completely reduce 20 ml of the Soxhlet solution.
Titration of sample. First, carry out an approximate titration: pipette 10 ml of solutions (a) and (b) into a flask and add 10 ml aliquot of-the sample solution. Add 40 ml of water and bring to boil. If blue colour persists, titrate with a standard working solution and calculate the approximate sugar content of the sample.
To accurately determine the sugar content, pipette 10 ml of Soxhlet solutions (a) and (b) into a flask and add an aliquot of the sample solution. The volume of sample used will depend on the sugar content of the sample (see Table 1).
Table 1 Sample Volumes Used in Soxhlet Titration
|
ml H2O |
ml sample |
g sample in aliquot |
Total sugar as invert, % |
|
40 |
10 |
0.08 |
73 |
|
35 |
15 |
0.12 |
82-58 |
|
30 |
20 |
0.16 |
61-41 |
|
25 |
25 |
0.20 |
49-35 |
|
20 |
30 |
0.24 |
41-29 |
(Reproduced from official Methods of Analysis of the AOAC, 1970)
Add water as indicated in the table, mix, and boil. During boiling, add a quantity of working standard from a burette so that the titration is nearly complete. Add methylene blue and complete the titration. Calculate the percentage sugar (as invert) by the formula:
% sugar = (F - M) × 1 × 100/W
where
F - is the volume of standard needed to reduce 20 ml of Soxhlet solution,
M - is the volume of standard sugar solution required to complete the back titration,
1 - is the weight of invert sugar in 1 ml of working standard, and
W - is the weight of sample in aliquot used.
4.7.2 Potassium
Reagents and equipment:
(a) hydrochloric acid (concentrated),(b) potassium standard
To prepare stock solution (500 ppm K), dissolve 0.477 g potassium chloride (Analar) and make up to 500 ml with distilled water. To prepare working standard (10 ppm), dilute 1:50.(c) silica crucibles,
(d) flame photometer, and
(e) muffle furnace.
Method:
Dry 2 g of sample in a silica crucible at 100°C to expel moisture. Add a few drops of pure olive oil and heat over flame until swelling stops. Ash at 500°C in muffle furnace for 24 h, cool, and add 2 ml concentrated hydrochloric acid to dissolve the residue. Make up to 100 ml. Take 1 ml of this solution and make a further dilution to 100 ml.
Set the flame photometer to give a reading of 100 with the 10 ppm standard, and then read sample solution. If the sample reading does not fall between 50 and 100 make a fresh dilution to give an appropriate reading.
4.8.1 Urease activity in soybean meal
Reagents:
(a) dimethylaminobenzaldehyde solution (DMAB)Dissolve 16 g DMAB in 1 litre 95% ethyl alcohol, and add 100 ml concentrated hydrochloric acid (stable for one month).(b) pyrophosphate buffer
Dissolve 23.3 g Na4P2O7 in approximately 980 ml distilled water. Add 3 ml of concentrated HCl and then additional HCl until the pH of the buffer is 7.7-7.8. Dilute to 1 litre.(c) buffered urea solution
Dissolve 04 g urea in 1 litre pyrophosphate buffer (stable for 1 week).(d) zinc acetate solution
Dissolve 22.0 g zinc acetate 2H2O in distilled water, add 3 ml of glacial acetate acid, and dilute to 100 ml.(e) potassium ferrocyanide solution
Dissolve 10.6 g K4Fe (CN)6, 3H2O in distilled water, and dilute to 100 ml.(f) charcoal.
Apparatus:
(a) water bath at 40°C, capable of maintaining temperature within ± 1°C, with shaking device,
(b) conical flasks, 125 ml,
(c) volumetric flasks, 25 ml, and
(d) spectrophotometer.
Method:
Accurately weigh 1 g of soybean meal into a conical flask and add 50 ml of the buffered urea solution. Incubate in water bath for exactly 30 min at 40 C with shaking. Remove from water bath and quickly add 0.5 ml each of concentrated HCl, ferrocyanide solution, zinc acetate solution, and 0.1 g of charcoal. Shake for 15 min and filter. If the filtrate is coloured, repeat the procedure using more charcoal. Pipette 10 ml aliquots of the filtrate and the DMAB solution into a 25 ml volumetric flask and make up to volume with distilled water. Make up also a reagent blank (10 ml DMAB made up to 25 ml with water) and a urea blank (10 ml buffered urea solution and 10 ml DMAB made up to 25 ml with water). Prepare a standard curve by pipetting aliquots of buffered urea solution from 2 to 12 ml into 25 ml volumetric flasks, adding 10 ml of DMAB and make up to volume.
Mix flasks well, stand in water bath at 25°C for 10 min, and then read at 430 mm. Calculate urease activity as mg/litre urea in urea blank less mg/litre urea in sample.
4.8.2 Free gossypol in cottonseed meal
Free procedures are described for the determination of free gossypol: the first for normal meals, and the second for meals which have been chemically treated and contain dianilinogossypol.
Reagents:
(a) aqueous acetone, 7 parts acetone, 3 parts distilled water (v/v);(b) Aqueous acetone - aniline solution
To 700 ml acetone and 300 ml distilled water add 0.5 ml redistilled aniline. Prepare solution daily.(c) Aqueous isopropyl alcohol solution: 8 parts isopropyl alcohol, 2 parts distilled water (v/v),
(d) Aniline
Distill reagent grade aniline over a small quantity of zinc dust, discarding the first and last 10 percent of the distillate. Store refrigerated in a brown glass stoppered bottle. Stable for several months.(e) Standard gossypol solution
(i) Dissolve 25 mg of pure gossypol in aniline-free acetone and transfer to a 250 ml volumetric flask using 100 ml of acetone. Add 75 ml of distilled water, dilute to volume with acetone, and-mix.(ii) Take 50 ml of solution (a), add 100 ml pure acetone, 60 ml of distilled water, mix, and dilute to 250 ml with pure acetone. Solution (b) contains 0.02 mg gossypol/ml and is stable for 24 h in darkness.
Apparatus:
(a) mechanical shaker,
(b) spectrophotometer,
(c) conical flasks, 250 ml capacity,
(d) volumetric flasks, 25 and 250 ml, and
(e) water bath (boiling).
Method:
Grind sample to pass 1 mm screen taking care not to overheat. Take approximately 1 g of the sample and add 25 ml of pure acetone. Stir for a few minutes, filter, and divide filtrate into two. To one portion add a pellet of sodium hydroxide and heat in a water bath for a few minutes. A light yellow extract which does not change colour with sodium hydroxide indicates that the cottonseed meal is untreated and procedure (1) should be used. A deep orange red colour in the tube containing sodium hydroxide indicates the presence of dianilinogossypol and this requires that procedure (2) be used.
Procedure (1): weigh 0.5 to 1 g of sample, depending on expected gossypol content, into a conical flask and add glass beads. Pipette in 50 ml of aqueous acetone solution, stopper the flask, and shake for one hour. Filter, discarding the first few ml of filtrate, and then pipette out duplicate aliquots into 25 ml volumetric flasks. (Take aliquots from 2 to 10 ml, again depending on expected gossypol content.) Dilute one of the aliquots to volume with aqueous isopropyl alcohol (Solution a) while to the other aliquot (Solution b) add 2 ml redistilled aniline; heat in a boiling water bath for 30 min together with a reagent blank containing 2 ml of aniline and a volume of aqueous acetone solution equal to the sample aliquot. Remove solution b and the blank, add sufficient aqueous isopropyl alcohol to effect homogeneous solution, and cool to room temperature in a water bath. Dilute to volume with aqueous isopropyl alcohol.
Read samples at 400 mu. Set instrument to 0 absorbance with aqueous isopropyl alcohol, and determine absorbance of solution a and reagent blank. If the reagent blank is below 0.022 absorbance proceed as below, otherwise repeat the analysis using freshly-distilled aniline.
Determine the absorbance of solution b, with the reagent blank set at 0 absorbance. Calculate the corrected absorbance of the sample aliquot: the corrected absorbance is the absorbance of solution b minus the absorbance of solution a. Determine the mg of free gossypol present in the sample solution using the calibration curve (see below).
Procedure (2): Weigh 1 g of sample into a conical flask, add 50 ml aqueous acetone, shake, and filter as above. Pipette duplicate aliquots of the filtrate (from 2 to 5 ml, depending on expected free gossypol level) into 25 ml volumetric flasks. Dilute one of the aliquots to volume (solution a) with aqueous isopropyl alcohol and leave for at least 30 min before reading on the spectrophotometer. Treat the other aliquot (solution b) as in procedure (1), determine the absorbances of solutions a and b as before, and calculate the apparent content of gossypol in both solutions a and b by using the calibration curve (see below).
Preparation of calibration curve: pipette duplicate 1, 2, 3, 4, 5, 7, 8, and 10 aliquots of the 0.02 mg/ml gossypol standard into 25 ml volumetric flasks. Dilute one set (solution a) to volume with aqueous isopropyl alcohol and determine absorbances as previously. To the other set (solution b) add 2 ml of redistilled aniline and proceed as previously. Prepare one reagent blank, using 2 ml aniline and 10 ml of aqueous acetone, heated together with the standards. Determine absorbances as in procedure (1) and calculate the corrected optical density for each standard solution:
Corrected absorbance = (absorbance solution b - absorbance solution a). Plot the standard curve, plotting corrected absorbance against gossypol concentrate in the 25 ml volume.
Calculate free gossypol percent in normal meals as:
where
G - is the graph reading
W - sample weight
V - aliquot volume used
For chemically treated meals:
where
A - mg apparent free gossypol in sample aliquot (a)
B - mg apparent free gossypol in sample aliquot (b)
W - sample weight
V - aliquot volume used
4.8.3 Thioglucoside determination
The method described will give approximate thioglucoside content but does not allow the individual thioglucosides and isothiocyanates to be determined.
Reagents and apparatus:
(a) barium chloride (5% solution),
(b) volumetric flasks, 600 ml, and
(c) steam bath.
Method:
To 10 g meal (de-fatted by Soxhlet extraction) add 250 ml distilled water, hydrolyse at 54°C for 1 h and then boil for 2h, keeping volume constant. Filter, retaining filtrate, and wash residue three times with 50 ml hot water. Add washings to initial filtrate and make up volume to 600 ml. Precipitate barium sulphate by heating and adding excess barium chloride solution. Leave on a steam bath for a few hours and then filter. Ash in a muffle furnace and then weigh precipitate.
Calculate approximate thioglucoside content as:
4.8.4 Aflatoxin analysis
A method of aflatoxin analysis is outlined below which is suitable for materials such as groundnut meal, coconut meal, and palm kernel meal. For full details of the method, and for alternative procedures reference should be made to Methods of Aflatoxin Analysis by B. D. Jones (1972), Report No. G70, Tropical Products Institute, London.
Reagents:
(a) chloroform (reagent grade),
(b) diethyl ether (reagent grade),
(c) chloroform/methanol mixture (95/5 v/v),
(d) "Celite", diatomaceous earth,
(e) Kieselgel 'G' (Merck),
(f) qualitative standardHelps to distinguish aflatoxin spots from other fluorescent spots which may be present. A groundnut meal containing aflatoxins B, obtainable from the Tropical Products Institute, London, can be used for this purpose.
Apparatus:
(a) thin layer chromatography plates, 20 X 20 cm,
(b) UV lamp, peak emission at 365,
(c) bottles, wide-mouthed, 250 ml,
(d) micropipettes, and
(e) shaking device.
Method:
Weigh 10 ml of material into a wide mouthed bottle and thoroughly mix in 10 ml of water. (If high fat material is used, a prior Soxhlet extraction with petroleum ether will be necessary.) Add 100 ml of chloroform, stopper with a chloroform resistant bung, and shake for 30 min. Filter the extract through "Celite", take 20 ml of filtrate and make up to 25 ml (solution a). Take another 20 ml of filtrate and concentrate to 5 ml (solution b).
Prepare thin layer plates by shaking Kieselgel 'G' (100 g) with water (220 ml) for 20 min and apply the mixture to the plates with a suitable apparatus to a depth of 509m. Leave for 1 h, then dry at 100°C. Spot 10 and 20m of solution b, and 5 and 10 m of solution a onto a plate, together with a qualitative standard spot, in a line 2 cm from the bottom of the plate and at least 2 cm in from each side. Carry out the spot application in subdued light.
Develop the plate in diethyl ether to a height of 12 cm. Allow to dry in subdued light then redevelop the plate in chloroform methanol (95/5, v/v) to a height of 10 cm from the baseline. Examine the plate in a dark room, 30 cm from the UV source. The presence of a blue fluorescent spot at Rf 0.5 to 0.55 indicates aflatoxin B (check that the standard spot also lies in this range). The presence of a second spot at Rf 0.45 to 5 indicates aflatoxin G. The toxicity level of a sample can then be classified in terms of aflatoxins B and G according to Table 2.
Table 2 Toxicity Levels for Alatoxins B and G
|
Vol. applied (m l) |
Concentration of aflatoxins (m g/kg) |
Toxicity level of fluorescence observed |
|
|
No fluorescence |
With fluorescence |
||
|
5m (soln. a) |
< 1000 |
> 1000 |
very high |
|
10m (soln. a) |
< 500 |
500-1000 |
high |
|
10m (soln. b) |
< 100 |
100-500 |
medium |
|
20m (soln. b) |
< 50 |
50-100 |
low |
Cockerell, I. and D. Holliday, 1975 Quality control in-the animal feedstuffs manufacturing industry. Trop.Prod.Inst.Rep., (G97)
Jones, B.D. 1972, Methods of aflatoxin analysis. Trop.Prod.Inst.Rep., (G70)
![]()
![]()
![]()