Chapter 2. Main soil contaminants and their fate in the soil environment
The chemical nature and properties of soil contaminants
Chemicals that act as environmental contaminants in soil and which potentially cause hazards, are either inorganic or organic compounds. Figure 1 introduces a systematic categorization of some of the most common contaminants in soils according to their chemical properties; as emerging contaminants can fall into a wide range of categorisations they are not included.
Figure 1. Systematic categorization of the main pollutants in soils according to IUPAC (Nič et al., 2009). Halogenated compounds comprise fluorinated, chlorinated, and brominated compounds.

The most commonly occurring inorganic soil contaminants are trace elements such as arsenic (As), cadmium (Cd), chromium (Cr), copper (Cu), mercury (Hg), lead (Pb), manganese (Mn), nickel (Ni), zinc (Zn), and radionuclides. Despite the natural occurrence of trace elements, hazard to the environment and human health can result if these elements are present at concentrations and/or in a chemical form that can be toxic for living organisms. Inorganic contaminants are by nature persistent and can occur in many different forms such as salts, oxides, sulphides, or organo-metallic complexes, or may be present in the form of ions dissolved in soil solution, depending on the soil pH. The partitioning among air, water and soil is driven by chemical processes such as adsorption to particles or pH-dependent dissolution in water. Unlike organic contaminants, which can be degraded when metabolized by different organisms, trace elements cannot be degraded by metabolic processes. However, an essential peculiarity of trace elements is the potential to exist as different species (e.g. different oxidation states as in chromiumIII / chromiumVI or arsenicIII / arsenicV) and tendency to form bioavailable metal-organic compounds (e.g. methylmercury or tetramethyl-lead), which effectively determines the bioavailability and toxicity. In practice, most assessments of trace elements measure and report total concentrations (e.g. of chromium) rather than the specific chemical form of the element (e.g. chromiumIII or chromiumVI) due to the complexity of analyses and interpretation of sequential extraction methods in laboratories. Additionally, integration of the physico-chemical interactions in the soil matrix with risk assessment approaches and soil contaminant quantifications is limited.
Soil pollution by radionuclides originates either from natural processes such as parent rock weathering and volcanic eruptions or from anthropogenic activities such as historical refinement of radium (for cancer treatment) and uranium, the use of radioactive phosphate and cobalt-ores, nuclear weapon tests or nuclear accidents. The key difference between pollution by trace elements and by radionuclides is that the adverse effects of trace elements are linked to chemical reactions taking place in living cells while radionuclides may damage living cells by emitted radiation caused by the radioactive decay. Consequently, different radionuclides are characterized by specific half-life1 times and the type of radiation.
In contrast, organic contaminants are carbon-based molecules, many of which are anthropogenic in origin, but also to a minor extent compounds derived from natural processes such as wildfires or volcanic eruptions. Synthetic organic contaminants may be produced for specific uses, such as pesticides (e.g. dichlorodiphenyltrichloroethane (DDT), dieldrin, and hexachlorobenzene (HCB), hexachlorohexane (HCH, lindane), and endosulfan) or as industrial chemicals or intermediate chemicals, such as polychlorinated biphenyls (PCBs) or other halogenated organics and volatile organics (VOCs) such as benzene, toluene, and chloroform. Organic contaminants may also be produced unintentionally as by-products such as industrial emissions, frequently from mining and petroleum industries, that release polycyclic aromatic hydrocarbons (PAHs). The PAHs are also formed naturally in small amounts by volcanic eruptions or forest fires (Kim, Choi and Chang, 2011; Kozak, 2017).
Some organic contaminants are volatile and many have low solubility in water resulting in high absorptive potential to lipids (lipophilicity), forming stable bonds with lipids and carbohydrates in organisms’ tissues and with soil organic matter (Meijer et al., 2003; Sweetman et al., 2005). Some organic contaminants (specifically some agrochemicals) are designed to be soluble in water and form water-soluble ions. Another environmentally relevant characteristic of many organic contaminants is persistence in the environment and potential toxicological effects. A contaminant is considered persistent if its half-life is at least of 60 to 120 days in water (sea water and fresh water) or 120 to 180 days in soils (ECHA, 2017). Highly toxic and persistent organic compounds, classified as persistent organic pollutants (POPs) according to Annex D criteria of the Stockholm Convention, have attracted the attention of the scientific community and policy sectors since the middle of the twentieth century and in particular since the Stockholm Convention was adopted on 22 May 2001.
The extent and duration of soil pollution and the risk of harm of a specific contaminant or contaminant class depends on several determinants:
- the chemical nature of the contaminant; that is, inorganic compounds versus organic compounds;
- the inherent physico-chemical properties, for example, chemical structure, volatility2, water solubility, lipophilicity3, lipophobicity;
- the inherent toxicity of the compounds in their various forms;
- parameters describing the interaction and transformation of the compound with environmental media and biological organisms such as solution partition coefficients, decay rate of radionuclides, metabolization (degradation) rates;
- the source of origin and pathway to the environment, for example, geogenic versus anthropogenic, direct release or disposal in soil versus atmospheric deposition;
- place and time of emission of contaminants;
- emission quantities;
- soil concentration and analytical detection values;
- bioavailability and/or toxicity of contaminants, as well as species (e.g. chromiumIII vs chromiumVI) or transformation products (e.g., DDT vs DDE), depending on environmental conditions (e.g. soil pH, moisture, weathering conditions);
- land use of and sensitivity to receptors in affected areas; for example, agricultural and forestry activities, recreational, residential or industrial areas.
2.2.1. Inorganic contaminants
This group of contaminants includes elements or compounds that occur naturally in parent rock or that have an anthropogenic origin. The major inorganic contaminants (trace elements, radionuclides, explosives and asbestos) are described below.
2.2.1.1. Trace elements
Trace elements4 (see Glossary for clarification) constitute inorganic compounds that are ubiquitous in nature. Some elements are essential micronutrients to soil microorganisms, plants, and animals, such as iron (Fe), copper (Cu), zinc (Zn), manganese (Mn), nickel (Ni), boron (B), selenium (Se) and molybdenum (Mo) (Viets, 1962), while other elements have no known metabolic function, such as lead (Pb), cadmium (Cd) and mercury (Hg) (Eisler, 2006; Flora, Gupta and Tiwari, 2012). The WHO (1996) differentiates between trace elements that are (i) essential (chromium, copper, molybdenum, selenium, zinc; (ii) probably essential (manganese, nickel, vanadium); and (iii) toxic elements (aluminium, arsenic, cadmium, lead, lithium, mercury, and tin).
Trace elements normally occur at small amounts in soils and plants. The total trace element content in specific areas depends on the geological parent material, the location, soil-forming processes that include weathering, leaching and erosion (which contribute to the mobilization and spatial distribution of trace elements in the environment) and anthropogenic sources such as fertilizers, sewage sludge, wastewater, industrial emissions, solid wastes, road dust, and atmospheric deposition (Huang et al., 2015). Trace element concentrations are expressed on a weight per unit weight basis e.g. micrograms or milligrams per kilogram of soil dry weight (µg/kg d.w. or mg/kg d.w.). The abundance of trace elements in soils may range from more than 1 000 mg/kg (e.g. aluminium, manganese, barium) to less than 1 mg/kg (e.g. cadmium, gold, mercury, molybdenum, selenium, silver) (Kabata-Pendias, 2010).
Anthropogenic activities such as mining, smelting, ore processing, gas works and any kind of metallurgical industry, sewage application, fertilizer production and use, and fossil fuel combustion for power generation, release trace elements into the environment. This occurs both from atmospheric emissions and due to direct release in soil and effluents (Cho et al., 2019; Hernandez et al., 2003; Hołtra, Anna & Zamorska-Wojdyła, 2020; Kabir et al., 2012). In addition to emission to air, the illegal dumping or dumping without proper containment of industrial waste poses a threat to the soil environment (Bailon et al., 2018; Hołtra, Anna & Zamorska-Wojdyła, 2020; Masindi and Muedi, 2019).
Arsenic, cadmium, chromium, copper, mercury, lead, manganese, nickel, and zinc are trace elements of high concern due to toxic effects at low concentrations to humans and other organisms, and due to the worldwide distribution (Driscoll et al., 2013; Hettelingh et al., 2015; Kabir et al., 2012; Li, Li and Jennings, 2018; Rahman and Singh, 2019). A detailed description of these trace elements, including the natural and anthropogenic sources and uses, is given in Table 1. Identifying the sources of trace elements in the environment is of key importance to understanding the pollution patterns and natural global cycles in addition to making decisions concerning soil pollution remediation (Cloquet et al., 2006).
Table 1. Natural and anthropogenic pollution sources of some common trace elements found as soil contaminants, including their uses.
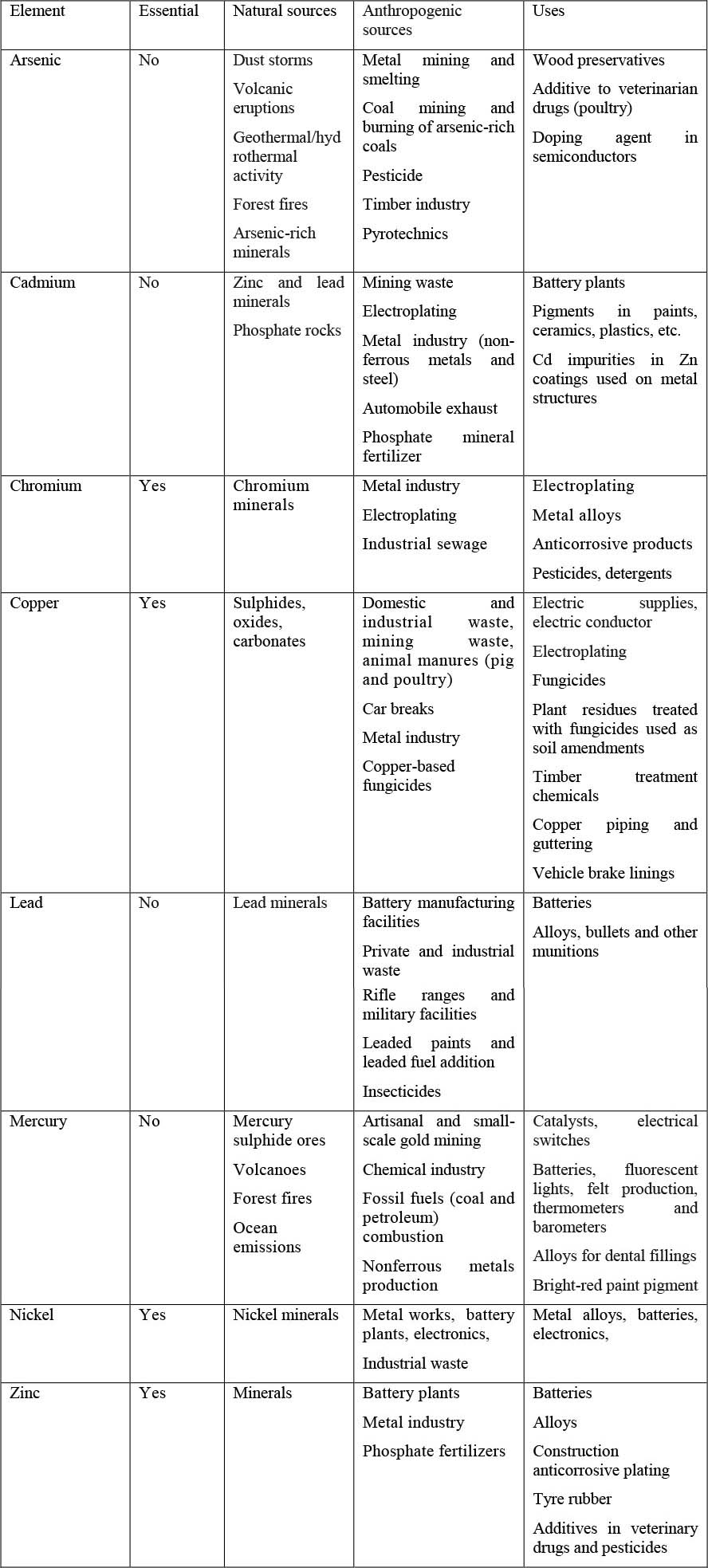
Arsenic
Arsenic is the twentieth most abundant element in the earth’s crust. Elevated concentrations of arsenic of natural origin are found in some regions in Asia and Africa. Arsenic can be mobilized through several chemical and biophysical processes: ion displacement; desorption (or limited sorption) at pH values above 8.5; reduction of arsenate to arsenite; and mineral dissolution, particularly reductive dissolution of iron and manganese (hydro)oxides, causing high arsenic concentrations in groundwater (Martiñá-prieto, Cancelo-gonzález and Barral, 2018; Pigna, Cavalca and Sommella, 2015). Unsafe use (e.g. untreated) of groundwater naturally contaminated with arsenic for irrigation or drinking water is causing health problems in the form of a disease known as arsenicosis in many areas of the world (Ahsan, DelValls and Blasco, 2009; Heikens, 2006).
Gold mining and quarrying activities and the chemical processes to extract gold are some of the anthropogenic sources of elevated concentrations of arsenic and mercury seen in parts of the African continent (Amonoo-Neizer, Nyamah and Bakiamoh, 1996). Arsenic is concentrated in the environment as a result of hydrothermal activities in many countries in Asia and the Pacific including China, Japan, Papua New Guinea and New Zealand (Garelick et al., 2008; Masuda, 2018).
Cadmium
Cadmium is a non-essential element that is naturally present in all soils (Smolders and Mertens, 2013) and is the seventh most toxic trace element (Jaishankar et al., 2014). Cadmium is naturally found in soil mainly via atmospheric deposition from volcano eruptions (Mulligan, Yong and Gibbs, 2001). Cadmium from these natural sources is usually complexed with the clay of shales as greenockite (CdS) or otavite (CdCO3) and is usually associated with zinc, lead or copper in sulphide form (Cameron, 1992).
As a by-product of zinc production, anthropogenic sources of cadmium to the environment are the main concern today, mainly in varied sources including alloys, polyvinyl chloride plastic (PVC) manufacture, solders, fungicides, enamels, motor oil, textile manufacturing, electroplating and rubber, sewage sludge and phosphate fertilizers (Matthews and Davis, 1984; Mulligan, Yong and Gibbs, 2001; Smolders and Mertens, 2013). Specifically, steel plating, pigment stabilization and nickel–cadmium batteries have been singled out as some of the main industrial sources (Mulligan, Yong and Gibbs, 2001).
Cadmium can affect ecosystem function at trace concentrations due to its toxicity (Smolders and Mertens, 2013). Cadmium is known to harm plant growth mainly because it competes and inhibits calcium, magnesium, phosphorus, potassium uptake and alters water uptake by plants (Nagajyoti, Lee and Sreekanth, 2010). Cadmium-affected plants show chlorosis, growth inhibition, browning of root tips and death (Guo et al., 2008; Sanità di Toppi and Gabbrielli, 1999).
Lead
Lead has been mined since ancient times for a great variety of purposes (Nriagu, 1983; Steinnes, 2013), and consequently, much of the observed lead in soil is sourced from anthropogenic emissions (Siccama and Smith, 1978).
Lead is naturally found in soils, mainly at deeper in the profile, from the alteration of the mineral galena and in smaller quantities from cerussite, anglesite, and crocoite (Mulligan, Yong and Gibbs, 2001). Lead can also be found in the surface layer of soil and in soil organic matter in large quantities from anthropogenic sources. Lead was widely used as an anti-knock agent in gasoline until banned in many countries from the 1990s (UNEP, 2019). Prior to the 1970s, lead was a common constituent of commercial and domestic paint and plumbing, and is often still present in older buildings due to these uses. The current primary sources of lead include lead–zinc smelting, ammunition, solder, glass, piping, insecticides and batteries (Steinnes, 2013; Yong and Mulligan, 2019). Lead is normally found in a divalent form and can replace calcium, strontium, barium and potassium in different soil minerals (Bradl, 2004) and soil organic matter is considered to be the main sink for lead. Under conditions of optimal plant growth, lead precipitates on root cell walls in an insoluble, amorphous form (Alloway, 2013).
Lead is the most commonly reported soil contaminant in the global database of contaminated sites - the Toxic Sites Identification Program (TSIP) - run by Pure Earth (Ericson et al., 2013; Pure Earth, 2019). Lead exposures result in over a million deaths annually—nearly 2 percent of global deaths according to the latest Global Burden of Disease Study by the Institute for Health Metrics and Evaluation (conducted in collaboration with WHO) (IHME, 2017). The global death rate has risen steadily every year since 2000 when most countries phased lead out of petrol (IHME, 2017). Ninety-two percent of deaths attributable to lead exposures occur in low- and middle-income countries. Exposures to lead are estimated to cost the economies of low- and middle-income countries nearly USD 1 trillion in GDP annually (Attina and Trasande, 2013).
Mercury
Mercury is the only trace element that occurs as liquid in its elemental form at room temperature. Mercury can easily evaporate and release to the atmosphere due to its high vapour pressure. Mercury can also form compounds which occur in gaseous and in solid particulate state, stimulating partitioning among air, water and soil. Soil and deep ocean waters act as a sink and a source of global mercury pollution respectively, with the balance between mercury deposition and volatilization controlled by partitioning among air, water and soil.
Mercury is a global contaminant that affects human and ecosystem health. The global burden of mercury pollution (Figure 2) is driven primarily by anthropogenic emissions, which greatly exceed natural geogenic sources. Artisanal and small scale mining, coal-fired power plants, nonferrous metals production and cement production are some of the major emitters of mercury (Bailon et al., 2018; NESCAUM, 2003; Prestbo and Gay, 2009; Sakata and Marumoto, 2005). Statistically-significant temporal trends and spatial patterns of deposition exist depending on regional and seasonal climate differences, namely decreasing wet deposition in line with increasing dry climate, and mercury deposition in summer is three times greater than that in winter (Prestbo & Gay, 2009). The global dispersion of gaseous mercury derived from natural and anthropogenic sources results in prolific deposition of mercury to land with volcanic emissions contributing about 20 to 40 percent of this flux (US EPA, 2014).
Figure 2. Global emissions of mercury given in tonnes per year.

Methylmercury is formed under reducing (that is, anoxic) conditions occurring in offshore and coastal marine environments, as well as freshwater and terrestrial ecosystems. This organic form is highly relevant from a toxicological point of view, because it readily bioaccumulates up the food chain. Humans and other animals are exposed to methylmercury by consumption of contaminated seafood (Driscoll et al., 2013). Another significant ingestion pathway affecting human health is the contamination of crops grown under anoxic conditions, for example uptake of methylmercury in rice in regions highly polluted by atmospheric deposition of mercury compounds (Xu et al., 2020).
Mercury use and production is now internationally regulated through the Minamata Convention on Mercury (Minamata Convention on Mercury, 2019).
2.2.1.2. Radionuclides
Radionuclides are present in soils from three sources. Primordial radionuclides remaining from the creation of the Earth typically have half-lives in the order of hundreds of millions of years. Examples include uranium-235, uranium-238, thorium-232, and potassium-40. These radionuclides end up in soil as part of natural soil-forming processes, such as weathering of rock by wind and water and breakdown by organic acids from living organisms. Radon is a radioactive, colorless, odorless, tasteless noble gas that is produced by the natural decay of radium, thorium and uranium. Cosmogenic radionuclides are continuously produced by bombardment of stable nuclides by cosmic rays, primarily in the atmosphere. The majority have shorter half-lives than the primordial radionuclides and include carbon-14, tritium-3, and beryllium-7; worldwide, cosmic radiation is the primary source of these radionuclides. The third source of radionuclides to soil is through anthropogenic activities, such as deposition from atmospheric testing of nuclear weapons and radiological events like the Chernobyl accident. Improper disposal of radioactive material also might contribute to contamination of soil with radionuclides. The main radionuclides of concern at polluted sites are strontium-90, cesium-137 and plutonium-239 with half-lives of 29 000, 30 000 and 24 000 years, respectively. Radiostrontium and radiocesium are readily taken up by plants, animals and humans due to their similarity to calcium and potassium, and are hence more toxic. Strontium can replace calcium and can accumulate in the bones and cause the development of dystrophic changes of the bone and articular system (Dutov and Yermolayev, 2013; Litvinov et al., 2008).
2.2.1.3. Asbestos
Asbestos is a generic term for a wide range of naturally occurring hydrated mineral silicate fibres belonging to the serpentine and amphibole groups of rock-forming minerals. Naturally occurring asbestos is mined and broken down into groups of loose fibres for commercial applications. The common types of asbestos available commercially are:
- - Chrysolite (white / hydrated magnesium silicate): the most commonly used worldwide and least hazardous
- - Amosite (brown or grey –/iron- magnesium silicate): medium hazard
- - Crocidolite (blue / sodium-iron silicate): most hazardous
Asbestos has been used in a variety of materials, such as in thermal insulation material, reinforcement of concrete/construction materials (fibro), fireproofing, brake linings, thermal, acoustic and chemical insulation (e.g. lagging, fire-rated doors, pipes, casing) (Frank and Joshi, 2014). Material that contains asbestos is referred to as friable or bonded. Friable asbestos material refers to any material that contains asbestos and is in the form of a powder or can be crumbled, pulverized or reduced to powder by hand pressure when dry. These products have significant potential for fibre production. Bonded asbestos material refers to any material that contains asbestos bonded with cement, resins or other similar materials.
Material containing asbestos is common in soils with current or historical older buildings/infrastructure, and asbestos must always be considered in contamination assessments for such sites (Wallis et al., 2020). Additionally, asbestos mining and refining sites are potentially significant sources of soil pollution.
All types of asbestos fibres are potentially harmful to human health; but effects depend on the type of asbestos material, its use, condition, location and exposure (Luus, 2007). Although no natural background values exists for asbestos in soils (Ander et al., 2013), it is estimated that 0.001 percent of asbestos in dry soil can exceed occupational exposure limits (0.1 fibres/ml air) (Langley, Gilbey and Kennedy, 2003). Inhaled asbestos causes a malignant tumour in lungs, abdomen or heart; this disease is called mesothelioma (Baumann et al., 2015; Molinari and Stevenson, 2020). Due to the high toxicity, many countries (67 as reported by the International Ban Asbestos Secretariat) have banned use of asbestos and asbestos containing materials (Kazan-Allen, 2019), but still major consumers of asbestos exist, such as India, China, Russia, Brazil and Indonesia, where populations are suffering from uncontrolled exposure (Chen, Sun and Wu, 2019).
It is worth mentioning that other asbestos-like minerals such as erionite, a naturally occurring fibrous mineral that belongs to a group of hydrated aluminosilicate minerals called zeolites found in certain volcanic tuffs, are also recognized as environmental contaminants and carcinogenic compounds (IARC Working Group on the Evaluation of Carcinogenic Risks to Humans, 2012).
2.2.2. Organic contaminants
2.2.2.1. Polycyclic aromatic hydrocarbons (PAHs)
Polycyclic aromatic hydrocarbons (PAHs) are compounds containing only carbon and hydrogen atoms organized in two or more annulated aromatic rings that are joined along shared edges. Terminal hydrogen atoms of the PAH backbone may be substituted by various side chains, and individual compounds (called homologues) show similar characteristics due to similar chemical structure. The conditions during reaction (such as the combustion temperature) determine the mix of homologues formed. Several thousand PAHs are possible in nature (National Research Council (US) Committee on Pyrene and Selected Analogues, 1983), being released by natural events such as volcano eruptions or forest fires or through a wide range of past and current anthropogenic activities such as production and combustion of petroleum and fossil fuels (Kim et al., 2013; Kozak, 2017; Ravindra, Sokhi and Van Grieken, 2008; Srogi, 2007). Polycyclic aromatic hydrocarbons are also produced during cooking of certain food items, such as grilled vegetables, toasted bread, and meat and smoked foods (Singh, Varshney and Agarwal, 2016).
Polycyclic aromatic hydrocarbons are solid at room temperature and are not soluble in water but are generally lipophilic; that is, the binding potential is strong to organic material, soil organic matter and fatty tissue, or dust particles. The properties of the individual PAH homologues depend on the number of hydrocarbon rings: the smaller the molecule, the smaller the lipophilicity and the higher the volatility. Consequently, PAHs partition in nature. Smaller PAH homologues show increased long-range-transport potential, and the larger and heavier homologues accumulate in organic matrices and the fatty tissue in living organisms including humans (Jones and de Voogt, 1999; Wilcke, 2000).
Many PAHs (specifically the larger homologues) are carcinogenic, mutagenic and/or toxic for reproduction. One of the most toxic is benzo(a)pyrene (Figure 3) with carcinogenic, mutagenic and teratogenic effects (Lawal, 2017). The United States of America Environment Protection Agency (US EPA) has identified 16 PAHs as Priority Pollutants which were selected as representative PAHs for regulation (Keith, 2015). The US EPA PAH list comprises naphthalene, acenaphthene, acenaphtylene, fluorene, phenanthrene, anthracene, fluoranthene, pyrene, benzo(a)anthracene, chrysene, benzo(b)fluoranthene, benzo(k)fluoranthene, benzo(a)pyrene, indeno[1,2,3-cd]pyrene, dibenzo(a,h)anthracene, benzo(g,h,i)perylene.
Figure 3. Molecular structure of benzo[a]pyrene.

2.2.2.2. Volatile organic compounds (VOCs)
The volatile organic compounds (VOCs) encompass a range of chemical classes (aliphatic5 and aromatic6) that exist primarily as liquids that are highly volatile or even gases at room temperature (Leo, Hansch and Elkins, 1971). Monocyclic aromatics commonly encountered in soils are the BTEX compounds (benzene, toluene, ethyl benzene and xylene) (Figure 4), which are easily biodegraded (Semple, Reid and Fermor, 2001). Representatives for halogenated aliphatic compounds are vinyl chloride, chloroform, trichloroethane (TCE) and tetrachloroethene (PCE). These compounds exhibit a range of toxic effects, with some promoting carcinogenic, mutagenic and teratogenic responses, and are not easily degradable but volatilization is an important loss process.
Figure 4. Molecular structure of BTEX contaminants.

Many VOCs are used widely as organic solvents in industry, for example in degreasing, dry cleaning, printing and painting operations, and are important contaminants at industrial sites. The BTEX compounds are associated with petroleum, especially gasolines, and are also used as solvents in many industrial processes.
The VOCs are one of the most significant categories of contaminants that are encountered at polluted sites, typically as mixtures (e.g. other hydrocarbons, BTEX and halogenated organic compounds). The volatility encompasses the difficulty in quantitative determination of the concentration in soil (Panagiotakis and Dermatas, 2015; Siegrist, 1991). Moreover, the volatility results in a high risk of fires and explosion. Benzene and carbon tetrachloride are two of the most toxic VOC compounds, both with carcinogenic effects, and both are commonly used solvents. On the basis of behaviour in soils and groundwater, hydrophobic organic liquids such as solvents are often grouped as non-aqueous phase liquids (NAPLs). Depending on the density with regard to water these compounds are subdivided into light non-aqueous phase liquids (LNAPLs) — which are less dense than water and float on water (e.g. BTEX) —, and dense non-aqueous phase liquids (DNAPLs) —which are denser than water and sink in water (e.g. tetrachloroethene).
2.2.2.3. Petroleum hydrocarbons (PHCs)
The term “petroleum hydrocarbons” is used to describe mixtures of organic compounds found in or derived from geological substances such as oil, bitumen and coal. This category of soil contaminants includes some mentioned above such as BTEX and PAHs, and other hydrocarbons with lower toxicological hazard (Canadian Council of Ministers of the Environment, 2001). These compounds have been considered as an independent group given the number and extent of soils polluted worldwide, and that remediation and management is similar. The PHCs are usually released into the soil environment due to accidental spills or leakages from oil storage containers, pipelines or vehicles (Balseiro-Romero, Monterroso and Casares, 2018).
Petroleum-derived products usually contain thousands of organic compounds, in varying proportions, formed by molecules of carbon and hydrogen, with smaller amounts of nitrogen, sulphur and oxygen, in forms of alkanes, cycloalkanes and aromatic compounds (Figure 5). PHCs possess a wide range of properties (volatility, solubility and toxicity), depending on molecular length, the aromaticity and polarity, which determine the fate and partitioning within the soil matrix (Balseiro-Romero, Monterroso and Casares, 2018). Smaller compounds such as BTEX are mainly present in the soil water fraction, due to increased solubility, or in the unsaturated pore space due to high volatility (e.g. benzene) (Berlin et al., 2015; Park and Park, 2010). In addition, longer chain hydrocarbons are less prone to biodegradation and therefore remain in the soil for longer periods, thus potentially posing a greater risk to human health and the environment (Shahsavari et al., 2017).
Figure 5. Molecular structure of some representatives of petroleum hydrocarbons.

Different processes occur that determine the fate of these contaminants in soil. Volatilization, dissolution and vertical migration, horizontal diffusion, natural attenuation including soil occlusion, chemical or microbial degradation and plant uptake are the main processes that control the fate and transport in soils (Balseiro-Romero, Monterroso and Casares, 2018).
2.2.2.4. Phenols, chlorobenzenes and chlorophenols
Phenols, chlorobenzenes and chlorophenols are chlorinated derivatives of benzene and phenol (Figure 6). Chlorobenzenes are cyclic aromatic compounds in which one or more hydrogen atoms of the benzene ring are substituted by a chlorine atom (Khatymov, Muftakhov and Mazunov, 2003). All of these compounds can cause toxic effects to humans and other organisms, and represent risk for ecosystems as potential precursors of dioxins (see PCDD/Fs) (Khatymov, Muftakhov and Mazunov, 2003). Phenol and related compounds can occur naturally in soil via synthesis by plants and fungi, release by decomposition of organic matter, or chlorination by microorganisms of mono and polyaromatic compounds in soil and water. Industrial activities represent important anthropogenic sources, such as the production of pesticides, dyes and pharmaceuticals (Malcom, Howe and Dobson, 2004; Michałowicz and Duda, 2007; Olaniran and Igbinosa, 2011). Highly chlorinated chlorobenzenes (HCB, PeCB) and pentachlorophenol (PCP) are listed in the Stockholm Convention Annex A for elimination of the production and use of certain chemicals (see POPs).
Figure 6. Molecular structures of phenol, chlorobenzene and chlorophenols.

2.2.2.5. Explosives
Modern explosives are nitrogen-containing organic compounds with the potential for self-oxidation with small gaseous molecules including nitrogen, water and carbon dioxide. Significant soil contaminants (including those in composition B, a commonly used military formulation) include 2,4,6-trinitrotoluene (TNT), hexahydro1,3,5-trinitro-1,3,5-triazine (RDX), and octahydro-1,3,5,7-tetranitro-1,3,5,7-tetrazocine (HMX) (Douglas et al., 2012). These compounds are recalcitrant to degradation, toxic and mutagenic (Kiiskila et al., 2015). It has been estimated that hundreds of explosives-contaminated sites exist within the United States of America, and even a greater number in Europe and Asia (Kalderis et al., 2011).
Production facilities and training ranges are the most common sites polluted with explosives, and the parent compounds and transformed products are sourced typically from artillery and mortar projectiles, hand grenades, and some small calibre ammunitions that permeate the soil and groundwater. Soil pollution can also result from the disposal of wastewater, which can be “red water” generated during the manufacturing of TNT or “pink” wash water associated with the load, assembly, and packing of materials in contact with TNT (Kalderis et al., 2011).
Besides explosive contaminants, military training sites are often also polluted with trace elements, mainly lead, due to bullets and their fragments, along with antimony, arsenic, copper, nickel, zinc, and silver (Islam et al., 2016).
2.2.2.6. Polychlorinated dibenzo-p-dioxins and dibenzofurans (PCDD/Fs)
Polychlorinated dibenzo-p-dioxins (PCDDs) are composed of two benzene rings joined by 2 adjacent oxygen bridges (C-O-C bonds), respectively, with varying degree of chlorine substitution on each benzene ring. Dibenzofurans (PCDFs) are similar with the benzene rings joined by one C-O-C bond and one direct bond without a bridging atom (C-C bond) (Figure 7). Between one and four of the H atoms on each ring structure can be substituted with chlorine atoms, enabling a total of 75 PCDD and 135 PCDF individual species. Each individual PCDD/F is referred to as a congener (WHO, 2000).
Figure 7. Molecular structure of PCDDs and PCDFs

Individual congeners present considerable variation in toxicity depending on the extent and positioning of chlorine substitution. The 2,3,7,8-substituted congeners are considered the most toxic to animals, and 2,3,7,8-TCDD is considered to be the most toxic member of this group, with reported mutagenic, carcinogenic and teratogenic effects (WHO, 2000). The degree of toxicity decreases with additional chlorination.
The formation of PCDDs and PCDFs occurs as a by-product of industrial processes associated with the chlorine and organochlorine industry, incomplete combustion during incineration processes including improper waste incineration, and burning of waste, and in particular electronic waste (Stockholm Convention, 2013; Weber et al., 2008). As a minor source, these compounds can be also formed during natural processes such as forest fires and volcanoes (Ferrario, Byrne and Cleverly, 2000; Kulkarni, Crespo and Afonso, 2008).
Although concentrations of PCDD/Fs in soils are low, the compounds are extremely persistent and hydrophobic, and can bioaccumulate in biota resulting in human exposure (Weber et al., 2018a). Currently a large proportion of the European population is considered exposed to greater that the tolerable intake values of the European Food Safety Authority, which has been set at 70 pg/kg bw/month (EFSA Panel on Contaminants in the Food Chain et al., 2018) and even low exposures can produce birth defects in small mammals, interact with DNA, and may cause carcinogenicity (Tuomisto, 2019).
2.2.2.7. Polychlorinated biphenyls (PCBs)
Polychlorinated biphenyls (PCBs) are a class of chlorinated man-made organic compounds comprising two benzene rings joined by a single C-C bond with varying degrees of chlorine substitution on the benzene rings (Figure 8). The molecular structure of a PCB allows ten positions on the biphenyl structure where chloric substitution may occur. This originates 10 possible homologue groups and 209 different congeners, which are named according to the number and position of chlorine atoms from PCB-001 to PCB-209 (Mills III, Thal and Barney, 2007). Depending on the conditions during PCB synthesis (e.g. reaction time, catalyst used), the degree of chlorination varies between 20 percent and 70 percent chlorine on a weight-by-weight basis, which in turn results in lower or higher abundances of highly chlorinated congeners while the relative abundance of the 209 possible congeners is determined by stereochemical effects7 (Faroon and Ruiz, 2016).
Figure 8. Molecular structure of polychlorinated biphenyls.

Studies have shown that a lower degree of chlorination results in higher volatility and lower lipophilicity. The PCB water solubility decreases with the degree of chlorination, although solubility is generally low. Due to the lipophilic nature, PCBs are absorbed by organic material or compounds (e.g. organic carbon in soils and sediments) and also in the adipose tissue of animals and humans. Specifically, the more highly chlorinated PCB congeners adsorb strongly to soil and sediment, and soil acts as a sink.
Due to high lipophilicity, low water solubility, and low biodegradability PCBs accumulate in the food chain and are rapidly absorbed from the gastrointestinal tract and distributed to and accumulate in the liver and adipose tissue. Because PCBs can cross the placenta and are absorbed in milk, they also accumulate in the foetus and infants, respectively (Faroon and Ruiz, 2016). The bioaccumulation, as extent of metabolism and degradation, strongly depends on the degree of chlorination and substitution pattern (Faroon et al., 2003).
Polychlorinated biphenyls were produced from 1929 to the 1990s. Because of unusually high chemical stability and electrical resistance, these compounds were used in a wide range of industrial applications including dielectric fluids in capacitors and transformers, hydraulic fluids, fire retardants and plasticizers. The PCBs were released throughout their lifecycle and are associated with environmental and human health impacts (Weber et al., 2018b). Given the concerns regarding environmental persistence and toxicity, PCB use and production was discontinued in most countries by the end of the 1970s, and PCBs belong to the group of 12 initial POPs under the Stockholm Convention. However, the legacy of PCBs already released into the environment and control of further releases remain a particular concern.
Dioxin-like polychlorinated biphenyls (dl-PCBs)
Some PCBs are identified as highly toxic, with effects are similar to dioxins. These are therefore called “dioxin like” or dl-PCBs. These PCBs show a similar structural relationship to dioxins and may bind to the aryl hydrocarbon receptor (Ah-receptor), a gene regulatory protein that induces the expression of target genes and mediates the biochemical and toxic responses to environmental contaminants (Bittner et al., 2006; Gasiewicz, 1997; Shen et al., 2008).
The more the positions of the chlorine atoms match the geometrical pattern of dioxins, the more the toxicity is similar to dioxins. Specifically, PCBs considered most biologically active and toxic have an almost co-planar structure like dioxins, notably 3,3’,4,4’,5-penta-CB (PCB-126) (Figure 9), followed by 3,3’,4,4’,5,5’ hexa-CB (PCB-169) and by 3,3’,4,4’-tetra-CB (PCB-077).
Figure 9. Molecular structure of PCB-126

2.2.2.8. Polybrominated diphenyl ethers (PBDEs)
Polybrominated diphenyl ethers (PBDEs) consist of two phenyl rings linked by an oxygen atom (Figure 10). The phenyl rings can be substituted by bromine atoms, to generate 209 possible PBDE congeners depending on number and position in the phenyl rings (EFSA, 2011). Since their introduction in 1970s, PBDEs have been used as brominated flame retardants (BFRs) in a broad array of polymeric materials such as plastics, foams, resins and adhesives (Besis and Samara, 2012; McGrath, Ball and Clarke, 2017). Global environmental contamination (including in polar regions) and toxic effects have been well documented, and despite replacement with novel brominated flame retardants (NBFRs), environmental issues related to risk for human health have not been solved (McGrath, Ball and Clarke, 2017). Novel brominated flame retardants have shown similar toxic patterns to PBDEs, and potential risk for humans and ecosystems remain due to contamination of soils.
Figure 10. Molecular structure of polybrominated diphenyl ether.

Commercial mixtures of different PBDE congener compounds were internationally banned and included in the Stockholm Convention (Besis and Samara, 2012; Stockholm Convention, 2021). Both PBDEs and NBFRs enter into the environment by atmospheric emission due to manufacturing, waste incineration, and recycling facilities, with electronic and electrical waste (e-waste) recycling being the greatest source (McGrath, Ball and Clarke, 2017). These compounds can contaminate soils by waste dumping, landfill leachates, application of sewage sludge and waste derived amendments. In soils, PBDEs and NBFRs can bind to organic matter, persist for long periods of time (estimated half-lives of 28 years), impact soil biota by bioaccumulation and biomagnification across the food chain, and be transferred to aquatic ecosystems (both fresh and marine waters) by sediments (Besis and Samara, 2012; McGrath, Ball and Clarke, 2017).
2.2.2.9. Perfluoroalkyl and polyfluoroalkyl substances (PFASs)
Perfluoroalkyl and polyfluoroalkyl substances (PFASs), especially the derived perfluoroalkyl acids (PFAAs), which include perfluoroalkyl carboxylic acids (PFCAs) and perfluoroalkyl sulfonic acids (PFSAs), are a family of synthetic molecules composed of a carbon chain that can be linear or branched, and partially or fully fluorinated (Figure 11) (Ghisi, Vamerali and Manzetti, 2019). Perfluoroalkyl and polyfluoroalkyl substances normally present high thermal, chemical and biochemical stability, low aqueous surface tension and amphiphilic behaviour, although a large number of homologues exist. The PFAS family may include between 5 000 and 10 000 different chemicals (US EPA, 2018) and therefore the range in behaviour highly diverse (Ghisi, Vamerali and Manzetti, 2019).
Figure 11. Molecular structures of the most common PFCAs and PFSAs.

Perfluoroalkyl and polyfluoroalkyl substances are persistent in the environment, toxic to both animals and humans, and highly mobile in aquatic environments, especially in the case of perfluorooctane sulfonate (PFOS) and perfluorooctanoic acid (PFOA), which are the most frequently detected PFASs (Banzhaf et al., 2017). Both of these compounds have been detected in surface- and ground-water worldwide, as well as in food (for example fish, meat, and eggs) resulting in concern for risk to human health and ecosystems (Banzhaf et al., 2017). Hepatotoxicity, tumour induction, developmental toxicity, immune-toxicity, neurotoxicity and endocrine disruption have been observed as possible negative effects (Ghisi, Vamerali and Manzetti, 2019).
The industrial, commercial and domestic applications for PFASs are multiple (Table 2), including as water repellent on clothing, leather, cookware and paper, as well as for decreasing surface tension in firefighting foam (Banzhaf et al., 2017; Ghisi, Vamerali and Manzetti, 2019). These substances can be found all over the world in all ecosystems, including remote parts of the globe: arctic regions and ecosystems in China, Europe and the United States of America are particularly affected (Banzhaf et al., 2017; Ghisi, Vamerali and Manzetti, 2019; Kucharzyk et al., 2017). Perfluorooctanoic acid, its salts and PFOA-related compounds and PFOS and its salts, and perfluorooctane sulfonyl fluoride (PFOSF) are listed in the Stockholm Convention Annex A and B, respectively.
Table 2. Major historical and current uses of polymeric and non-polymeric per- and polyfluoroalkyl substances (PFASs).


2.2.2.10. Pesticides
The term “pesticide” includes active substances, or mixtures of substances, used for preventing, destroying or controlling any pest causing harm to humans or interfering with human activities such as production, processing, storage, transport or marketing of food, wood and wood products, fibres and other agricultural commodities (FAO and WHO, 2016; Rodríguez Eugenio, McLaughlin and Pennock, 2018). Pesticides are also used in the public health sector as a vector control for diseases such as malaria (WHO and FAO, 2019). Today, there are about 1 000 different pesticides in use composed of more than 800 active ingredients and the number is still increasing (Sánchez-Bayo, 2011; Zhang, 2018). Insecticides, fungicides, nematicides, rodenticides and herbicides are some examples, which can also include insect repellents and insect or plant growth regulators (FAO and WHO, 2016). Different classifications of pesticides have been proposed by different organizations (e.g. WHO, Compendium of Pesticide Common Names (Wood, 2020)), depending on chemical composition and the chemical families to which they belong (e.g. organochlorines, organophosphates, organo-metals), or on toxicological effects, for example Acute Toxicity Hazard Categories from experimental data (Khan and Rahman, 2017).
Since the Second World War, when DDT and its insecticidal properties were developed, synthetic pesticides have been extensively used and released into the environment. The use of pesticides accelerated in particular in the 1950s and 1960s during the “Green Revolution8” in agriculture. However, the excessive and improper use of pesticides causes unwanted damage to non-target species, while persistence in the environment and toxic residues can impact beneficial and non-target organisms including humans, and can contaminate waters and soils on a global scale (including in remote areas such as the polar regions) over long time periods (Khan and Rahman, 2017). A number of pesticides and pesticide residues can bioaccumulate in high concentration in plants and animals and cause biomagnification9 in food chains (Stanley and Preetha, 2016).
Some pesticides are listed as persistent organic contaminants (POPs) by the Stockholm Convention (Stockholm Convention, 2021) and have a major relevance in the context of soil pollution being persistent, show potential for long-range transport, and cause adverse effects to environment and human health. The pesticides listed in the Stockholm convention are chlorinated organic compounds, such as aldrin, dieldrin, endrin, chlordane, DDT, heptachlor, hexachlorobenzene, mirex, toxaphene; hexchlorcaclohexane/lindane, endosulfan.
DDT
DDT and its metabolites are among the mostly discussed and regulated pesticides and POPs. DDT was invented by the Swiss chemist, Paul Herrmann Müller in 1940, and has been used intensively worldwide as an insecticide, in particular to combat the malaria-transmitting mosquito. The yearly production rate increased rapidly from some 10 000 tonnes/year to reach its estimated maximum of 150 000 tonnes/year in the 1960s (Schenker, Scheringer and Hungerbühler, 2008; Semeena and Lammel, 2003). Since the 1970s, DDT has been banned in many countries due to its environmental impacts, and its emission by production and use has significantly declined.
Technical DDT marketed as “DDT” mainly consists of p,p′-DDT (ca. 80−85 percent) and the isomer o,p′-DDT (ca. 10−15 percent) and only low amounts of p,p´-DDE (Spencer and Cliath, 1972). In nature, the two isomers p,p′-DDT and o,p′-DDT are metabolized aerobically and anaerobically and produce p,p′-DDE and o,p′-DDE, or p,p′-DDD and o,p′-DDD, respectively (Figure 12). the half-life decreases between the three compounds, being about 1000 days for DDT, 500 days for DDD and less than 10 days in soil, water and air for DDE, respectively (Schenker, Scheringer and Hungerbühler, 2008; Wegmann et al., 2004).
Figure 12. Molecular structures of DDT and its metabolites.

2.2.2.11. Persistent organic pollutants (POPs)
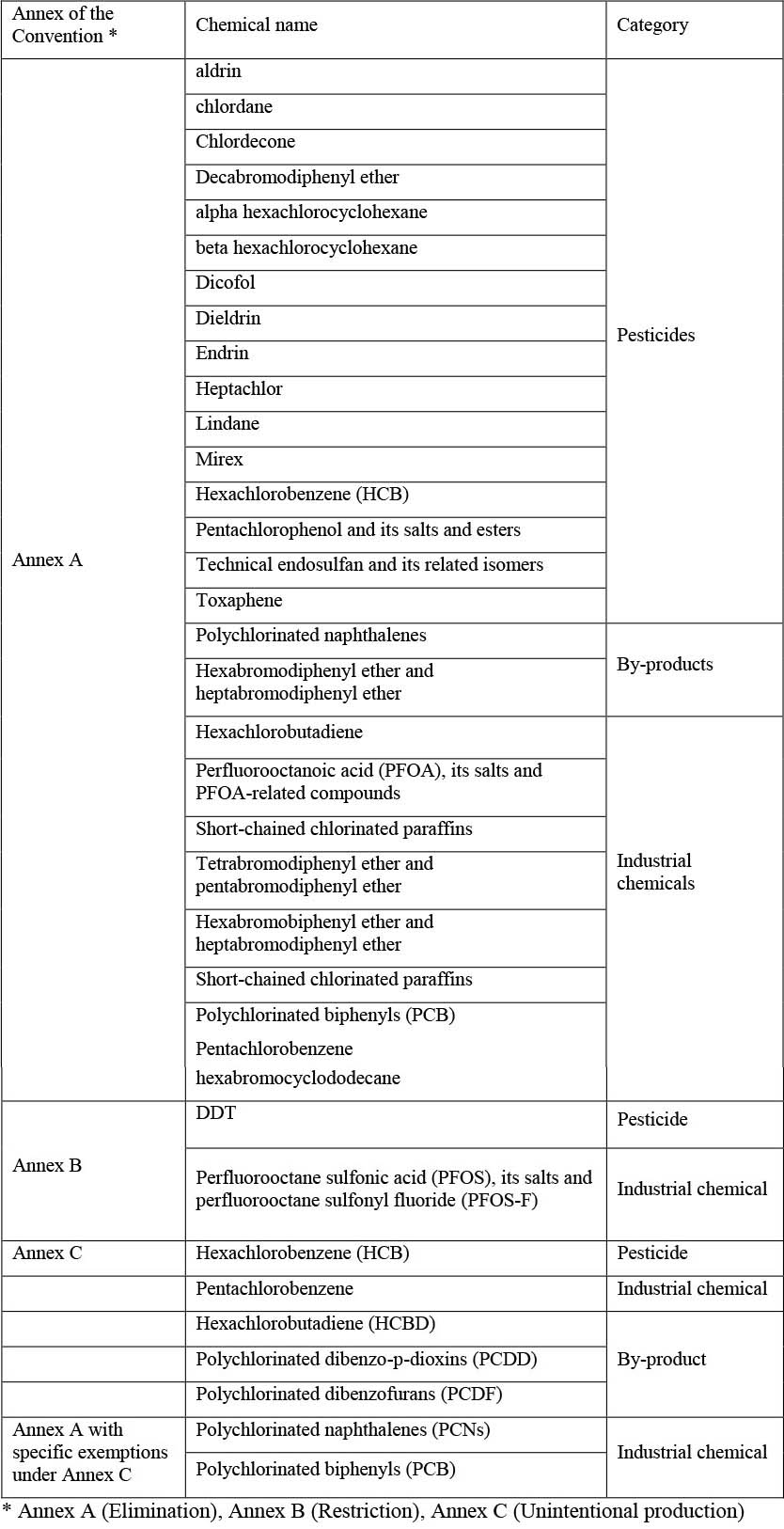
Persistent organic pollutants are organic chemicals which, due to their physical-chemical properties, are resistant to degradation in the environment (years to decades), exhibit a pronounced binding potential to organic material, including fatty tissues, and exhibit long-range transport potential through air, water or via body burden in migratory species. These pollutants include some of the contaminants mentioned above (e.g. some pesticides, PCBs or PFAS), but have been regulated by the international community because of concerns regarding toxicity and persistence in the environment. The use and production of POPs are internationally regulated by the Stockholm Convention on Persistent Organic Pollutants (see Chapter 5 for details about the Convention), which at the time of writing this report regulates 33 POPs (Stockholm Convention, 2021) (Table 3).
Table 3. The chemicals targeted by the Stockholm Convention, the Annex in which they are included and the major category they are classified into. Source (Stockholm Convention, 2021).
Solid scientific evidence exists regarding the significant toxicity of some POPs, which are transported across international boundaries, deposited far from the place of release due to the long-range transport, and bioaccumulate in terrestrial and aquatic ecosystems (Bhardwaj et al., 2018; Harley et al., 2019; Jones and de Voogt, 1999; Kallenborn, 2006; Nøst et al., 2013; Ren et al., 2017). Soils can act as sinks for POPs, due mainly to the high affinity to bind to organic carbon in soils and recalcitrance to degradation (Meijer et al., 2003; Posada-Baquero and Ortega-Calvo, 2011).
Persistent organic pollutants are detected in flora, fauna and wildlife and also in humans. Studies on human milk samples demonstrate POP residues in populations of industrial regions in central Europe (Croes et al., 2012), and even in remote regions far from emission sources (Kallenborn, 2006; Lohmann et al., 2007). The POPs are also detected in the general population, as demonstrated by the WHO/UNEP human milk study (UNEP, 2013). Possible harmful effects on organisms range from neurotoxic to teratogenic, carcinogenic, and endocrine disruptive effects. This is especially concerning because the lipophilic properties of these substances favour storage in the fat tissue of organisms, which consequently leads to biomagnification through the food chain (Jones and de Voogt, 1999).
2.2.3. Emerging contaminants
The classification of emerging contaminants refers to a broad group of synthetic or naturally occurring chemicals and microorganisms that have not previously been monitored in the environment, but due to potential toxicity or risk to human and environmental health are generating increasing concern among the scientific community and policy makers (Rosenfeld and Feng, 2011). Some of the contaminants cited here belong to the categories of organic and inorganic contaminants mentioned above, but because of emerging concern and limited understanding of fate and effects in soils have been described here as a separate category in this report for clarity and awareness.
2.2.3.1. Pharmaceuticals and personal care products (PPCPs)
Pharmaceuticals can be defined as any active chemical (natural or synthesized) that is designed to prevent or cure diseases and to improve quality of life for humans or animals (Ebele, Abou-Elwafa Abdallah and Harrad, 2017; Tijani et al., 2016). Pharmaceuticals are characterized by different chemical structures, applications and metabolism in humans and animals. According to the therapeutic uses, some of the most important of these compounds are antibiotics, anti-diabetics, anti-epileptics, anti-inflammatories and analgesics, antiulcer and antihistamine drugs, anticancer drugs, anti-anxiety/hypnotic agents, antidepressants, tranquilizers, lipid regulators, lipid-lowering drugs, antipyretics and anticonvulsants (Liu and Wong, 2013; Tijani et al., 2016; Yang et al., 2017b).
Pharmaceuticals are not completely assimilated by humans or animals; the original compound or partially metabolized (sometimes activated) drug residues are excreted. These compounds cannot always be completely degraded by conventional wastewater treatment plants before entering the environment (Baker and Kasprzyk-Hordern, 2013; Caliman and Gavrilescu, 2009; Sui et al., 2015). However, some research and practical evaluations of additional treatment processes to remove pharmaceuticals have started recently in Europe (Langenhoff et al., 2013; Wang and Wang, 2016). Pharmaceuticals are not included in the group of POPs; however, some compounds (e.g. carbamazepine) or metabolites of compounds are persistent. In addition, many PPCPs are not persistent because of chemical structure, but due to the continuous use and release into the environment are considered ‘pseudo-persistent’.
Personal care products are used to improve the quality of daily life. These compounds include antimicrobial agents (e.g. triclosan and triclocarban), fragrances (e.g. nitro musk), active ingredients of insect repellents (e.g. N,N-diethyl-m-toluamide (DEET)), sunscreen UV filters (e.g. 4-methyl-benzylidene-camphor (4-MBC)) and preservatives (e.g. parabens) (Ebele, Abou-Elwafa Abdallah and Harrad, 2017; Liu and Wong, 2013; Yang et al., 2017b). Most are bioaccumulative and persistent in the environment due to the lipophilicity (Caliman and Gavrilescu, 2009). To date, information on occurrence in the environment is incomplete, in addition to knowledge on effects on non-target organisms and potential risk of exposure (Tijani et al., 2016; Wang and Wang, 2016).
Pharmaceuticals and personal care products represent an unintentional and continuous release of substances into the environment. Currently, more than 5 000 pharmaceuticals are available worldwide (Van Doorslaer et al., 2014). The most important countries for pharmaceutical production are Brazil, Russia, India, South Africa and China. Global production is estimated in the range 100 000–200 000 tonnes/year (Tijani et al., 2016). Few PPCPs are monitored and regulated, and for most no legal requirements exist to assess the impact of long-term exposure (Caliman and Gavrilescu, 2009; Sui et al., 2015).
Pharmaceuticals and personal care products are released into the environment by multiple pathways, including municipal wastewater, domestic (private) wastewater (e.g. septic systems or uncontrolled releases from home of expired medicines), landfill sites, industrial effluents, hospital effluent, and land application of municipal sewage or domestic sewage as part of agricultural production, resulting in PCPPs becoming ubiquitous in water bodies (Sui et al., 2015; Tijani et al., 2016; Wang and Wang, 2016; Yang et al., 2017b). These compounds can accumulate in sediments and soils, leach to groundwater and contaminate drinking water (Yang et al., 2017b).
Concern is increasing regarding PPCPs persistence, mobility, toxicity, bioactivity and bioaccumulation (Caliman and Gavrilescu, 2009; Ebele, Abou-Elwafa Abdallah and Harrad, 2017; Sui et al., 2012; Yang et al., 2017b). The majority of pharmaceuticals are water-soluble and with a short life span, although some (e.g. sulfamethoxazole) can remain stable in the environment for more than a year (Tijani et al., 2016). These substances can induce physiological adverse effects at low doses, with potential health risks for humans, aquatic and soil organisms and ecosystem functionality in different environmental matrices (water, soil and sediment) (Ebele, Abou-Elwafa Abdallah and Harrad, 2017; Sui et al., 2012; Tijani et al., 2016). The adverse effects of pharmaceutical exposure include acute and chronic toxicity, bioaccumulation, disruptions of the endocrine system and drug-resistant pathogens (Figure 13) (Caliman and Gavrilescu, 2009; Rodríguez Eugenio, McLaughlin and Pennock, 2018; Sui et al., 2015). The fate of these compounds in soils is complex, analysis difficult, and typically knowledge of fate and behaviour in the available literature is limited.
Figure 13. Main mechanisms of antimicrobial-resistance.

2.2.3.2. Plastics and synthetic polymers
Plastics are widely used in nearly all aspects of everyday life because of versatility, high performance, high resistance and cost effectiveness. Elastomers, rubber-like polymers materials, share similar environment-related issues and health impacts (Andrady, 2003).
Polymers consist of very large macromolecules, characterized by average molecular weights of millions of grams per mole (from 1 000 to 10 000 atoms per molecule), and a long repetitive chain-like molecular architecture (Andrady, 2015). This class of unique giant linear molecules possess chemical and mechanical properties for multi-purpose applications in many fields, competing well with other materials such as glass, metal and wood for the same application (Andrady, 2015, 2003).
Plastics, polymers and other related materials mainly originate from the processing of crude oil and natural gas, and in some cases from other raw materials (such as coal and biomass). Studies of plastic occurrence in soil media usually consider the most common types, such as polyethylene, polypropylene, polyvinylchloride, polystyrene and thermoplastic polyester. Nevertheless, thousands of different plastics exist for a multitude of applications (Andrady, 2003). Two general groups of plastics exist, the thermoplastics and the thermosets. The first, which includes polyvinylchloride (PVC), polyethylene, polymethylmethacrylate, nylon and polycarbonate, are plastic materials that can be shaped several times by the application of heat and pressure. This group can be recycled by melting and used again for different applications and in different shapes. The second group, which includes epoxy and polyurethane, cannot be softened by heating, and are generally used for disposable applications without recycling (Andrady, 2003).
Polyethylenes consist of several copolymers of ethylene and represent the most widely used class of plastics worldwide (Andrady, 2003). These compounds are extensively used for packaging films (e.g. grocery bags). The second most widely used plastic resin is PVC, which is frequently used in plumbing pipes and window and door profilews. When incorporated with plasticizers such as organic phthalates, this resin is a very versatile and soft material, which can be used for packaging film and cable covering. Polypropylene is manufactured by direct polymerization of propylene, and can be degraded by light and heat without stabilizers and additives. Polyethylene terephthalate (PET) is a transparent resin widely used for bottles and jars and for packaging application due to stiffness, strength and temperature performance. Polystyrene is frequently used to give flexibility and water resistance to textiles and fibres (Andrady, 2015, 2003; Li, Tse and Fok, 2016).
The annual worldwide production of polymeric resins has grown from 1.5 million of tonnes in the 1950s to 299 million tonnes in 2013, and it is estimated to grow to nearly 500 million by 2100 (Li, Tse and Fok, 2016; Andrady, 2003). Consequently, plastic items and residues are commonly found in all environmental compartments and can be ingested by organisms, causing damage and possibly mortality, but may also be transferred across the food chain (Akindele, Ehlers and Koop, 2019; Barnes, 2019; Huerta-Lwanga et al., 2017; Hurley and Nizzetto, 2018; Li, Tse and Fok, 2016; Andrady, 2003). The fugitive emission of potentially toxic solvents, eluents and other chemicals (e.g. monomers) during plastic production, the use of toxic catalysts, such as chromium (IV) oxide-based catalyst in production, and particulate emissions can also represent an important burden of pollution, impairing human health and ecosystems.
Microplastics
Due to the high resistance to degradation and improper disposal, plastics can reach water bodies and soils and persist in the environment for decades (Li, Tse and Fok, 2016). Plastics can be partially broken down by mechanical action (agricultural machinery, garbage collection trucks and treatment plants) or weathering, including UV-radiation of sunlight, which leads to a great variety of plastics with different dimensions in ecosystems. Macroplastics are generally defined as having a size >5 mm, whereas microplastics refers to particles < 5 mm (Li, Tse and Fok, 2016; McCormick et al., 2014).
Microplastics can be formed by the chemical and physical weathering of plastics (secondary microplastics) or have a primary source (e.g. cosmetic beads, scrubbers). Due to the small size, microplastics are not completely removed by wastewater facilities, and are easily dispersed into the environment (Filella, 2015). Microplastics can absorb and subsequently leach toxic compounds such as trace metals, PCBs, PBDEs, nonylphenols and pesticides, increasing the burden of pollution (Gallo et al., 2018; McCormick et al., 2014).
2.2.3.3. Plasticizers
Plasticizers including bisphenol A (BPA) and phthalates, diesters of phthalic acid (Figure 14), are recognised endocrine disruptors causing diverse toxic effects, such as cancer and osteoporosis (Benjamin et al., 2015, 2017; Meeker, Sathyanarayana and Swan, 2009; Tran et al., 2015). These are a group of synthetic chemicals widely used in industry as plastic additives to make plastic goods more flexible (Benjamin et al., 2017; Meeker, Sathyanarayana and Swan, 2009).
Figure 14. Molecular structure of phthalate and BPA.

BPA is the most commonly used bisphenol and is mainly used to produce polycarbonate and epoxy resins. BPS is present in multiple electronic devices, sport equipment, medical devices, food storage containers, reusable bottles, building and construction materials, automotive parts, and in thermal papers, such as credit card slips, bank receipts, and fax papers (Chen, Chang and Minatoya, 2020). BPA can be released into the environment during production, manufacture of BPA-containing products, use of these products and disposal (Corrales et al., 2015). BPA has moderately water solubility, a low vapour pressure and high melting point, and is partially lipophilic. BPA has a short half-life of 4.5 days in water and soil and less than 1 day in air (Cousins et al., 2002); however, BPA has been found in all environmental matrices and in aquatic and terrestrial organisms, including humans (Chen, Chang and Minatoya, 2020; Corrales et al., 2015).
The first phthalate introduced was diethylhexyl phthalate (DEHP), in the late 1920s. Since 1931, phthalates have been also employed in the cosmetics and perfumery industries (Benjamin et al., 2017). The plasticizers are not chemically bound to plastic polymers, and consequently can easily leach into the environment and are released during plastic weathering. Due to widespread use, these compounds are widespread in the environment, including soils, and pose risks to human health and ecosystems (He et al., 2015; Net et al., 2015).
2.2.3.4. Manufactured nanomaterials (MNM)
Nanomaterials are defined as materials with at least one dimension between 1 and 100 nm, but the nanoparticles with all three dimensions between 1 to 100 nm are particularly important (ISO, 2008). Nanomaterials possess enhanced and unique properties (Ju-Nam and Lead, 2008), and thousands of consumer applications exist due to the exponential growth and development of new manufactured (or engineered/man made) nanomaterials (MNM), including plastics, electronics, textiles, cosmetics, catalysts and medicinal science (Klaine et al., 2008; Lead et al., 2018), aside from natural and incidental production (Figure 15).
Figure 15. Illustration of different types of nanomaterials and their possible sources.

The MNMs generally fall into seven classes: carbon based materials such as carbon nanotubes and fullerenes, metal oxides such as zinc oxide, titanium oxide in sunscreens, cosmetics and solar cells, semiconductor nanocrystals (quantum dots – cadmium sulfide, cadmium selenide), zero-valent metals including iron, gold and silver the greatest consumer application because of the antimicrobial activity, nanopolymers such as dendrimers, nanoclays and emulsions (Batley, Kirby and McLaughlin, 2013). The products, which are continually expanding, include nanoparticles as well as nanofibers, nanowires and nanosheets (Figure 15) (Saleh, 2020).
The MNMs can be applied deliberately to soil for remediation of pollution but are also released unintentionally through various pathways including wastewater and sludge (Kuenen et al., 2020; Pan and Xing, 2012). Soil is a major sink, and the current growth in nanotechnology for targeted pesticide and fertilizer delivery in food crop production will likely increase MNM input to soil systems (Lead et al., 2018). The different patterns of production, use and disposal of MNM in each country determine release into the environment, with sludge input to agricultural soils being one of the main sources of MNM pollution in soils (Kuenen et al., 2020). There is a growing consensus that MNMs are less toxic than equivalent dissolved materials but more toxic than the corresponding bulk materials (Lead et al., 2018). Nevertheless, with nanospecific aspects to fate, behaviour, bioavailability and toxicity in soils, and a scarcity in soil field based studies and knowledge on environmental concentrations, integration of the emerging nanotechnologies with improved risk assessment remains critical for environment and human health protection (Lead et al., 2018).
2.3 Soil properties that determine the fate and transport of soil contaminants
Contaminants enter the soil environment from different sources (see Chapter 3). Once in the soil, contaminants can move through the soil matrix relatively freely and show accumulation, transformation and degradation through chemical and biological processes (Figure 16). A soil is polluted when its function of buffering and filtering contaminants is exceeded due to the presence of contaminants whose nature, location or quantity produces undesirable effects on the environment or human health (UN Statistical Division, 1997, see the glossary on Chapter 1). However, the value that delimits this excess is different for each soil, each contaminant and each particular situation (e.g. different climatic conditions or mixtures of contaminants present). Soil properties will therefore affect the fate and transport of soil contaminants.
Figure 16. Routes of entrance and fate of contaminants in soils.

The fate and transport of contaminants within the soil depends on their partitioning among the solid, liquid and gas soil phases, which is determined by:
- physical interactions: trapping in soil micropores, occlusion in soil aggregates, transport with colloids in the soil solution;
- chemical interactions and transformations: adsorption onto charged surfaces of clay and soil organic matter, or dissolution into soil solution with pH-dependent effects on solubility as affected by chemical hydrolysis, oxidation-reduction reactions;
- biological interactions: mineralization or other transformations by soil organisms, transport by soil organisms through macropores and channels, and uptake by plant roots and transfer into root, shoot, leaf and fruiting tissues (which can be removed when crop plants are harvested);
- interactions between contaminants: contaminants can have synergistic or antagonistic effects when present simultaneously in the soil matrix;
- interactions among i), ii), iii) and iv) and ambient environmental conditions: for example, volatilization into soil gas at higher temperatures, greater dissolution into soil solution with higher soil moisture content, higher rates of microbial degradation under higher soil temperature and moisture, and photolytic degradation by sunlight.
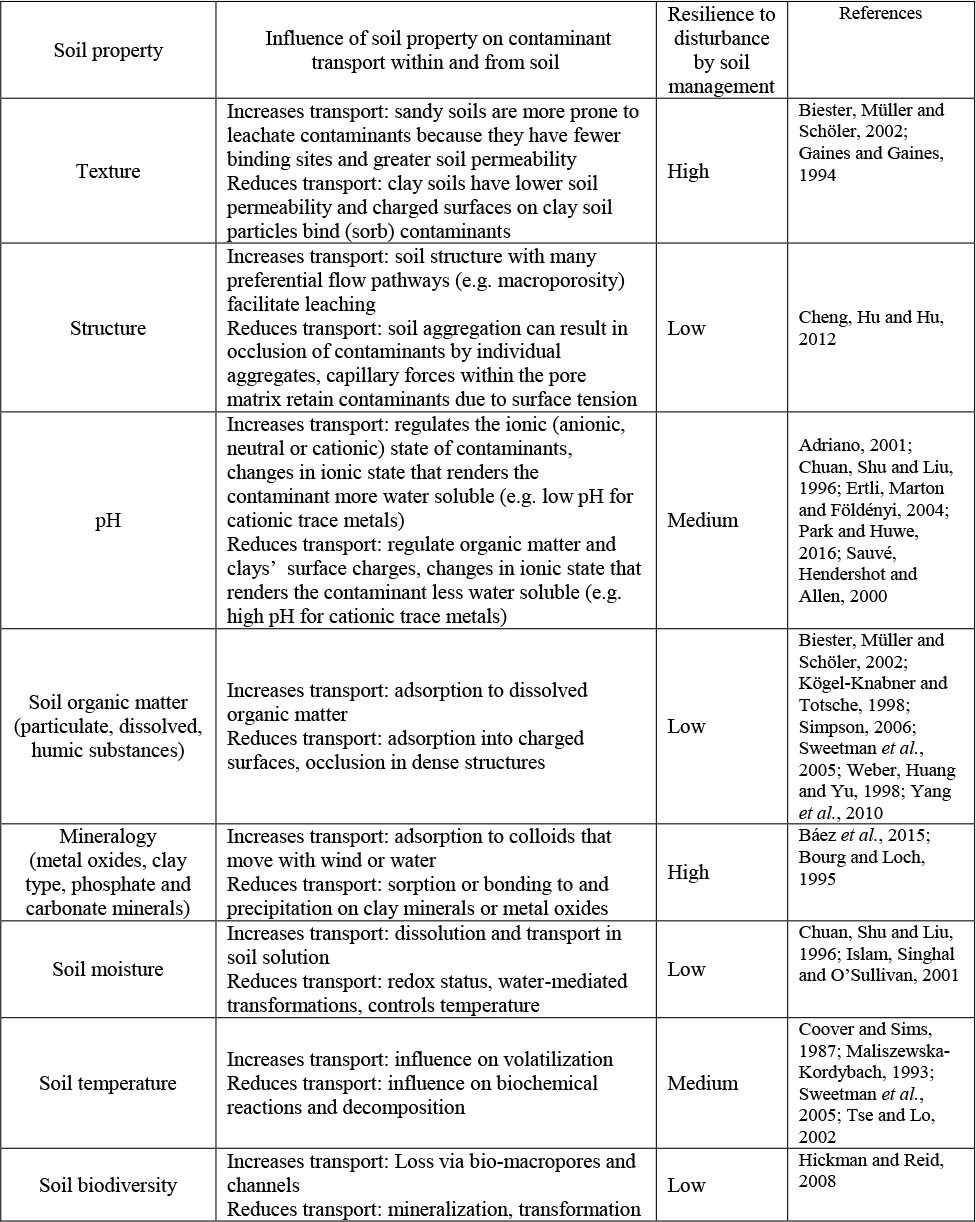
The occurrence and extent of these interactions is influenced by soil properties. The soil properties that determine the soil’s capacity to retain and accumulate contaminants are mainly soil pH, type and amount of organic matter, content of (hydro-)oxides of Fe, Al and Mn, type and proportion of clay particles, soil biodiversity and soil moisture (Table 4). The influence of these soil properties on contaminant transport does not operate in isolation: one or more properties interact and exert additive, synergistic or antagonistic effects on contaminant loss via volatilization, wind or water erosion, or leaching. The ultimate movement of contaminants in soils depends on the rates and thermodynamic parameters of the retention, infiltration, evaporation, dissolution and biodegradation of the contaminants, which determine partitioning and accumulation in the different components of the soil matrix and the environmental compartments (Breus and Mishchenko, 2006).
Soil components (clays, (hydro-)oxides of iron, manganese and aluminium, and organic matter) present negative and positive charges that can be permanent (inherent) or variable (depending on the pH and salt content of the soil solution by dissociation of active groups and association of hydrogen ions) (Osman, 2013; Sumner and Miller, 1996). Cations and cationic substances, and anions and anionic substances can bind to these charged surfaces. The exchanges between soil particle surface areas and soil solution are osmotically driven, controlled by sorption and desorption processes; these exchange sites determine the cation exchange capacity (CEC) and anion exchange capacity (AEC) of the soil. Yaron and co-workers present a detailed description of sorption mechanisms. In this chapter, those mechanisms are not described in detail but are mentioned when relevant (Yaron, Dror and Berkowitz, 2012).
Partitioning among different media usually follows an equilibrium between the contaminant sorbed on solid surfaces and the contaminant dissolved in liquid (or gaseous) phases, controlled by the chemical characteristics of the contaminant (e.g. hydrophobicity, volatility). Several distribution coefficients have been developed over the years (e.g. partitioning coefficient between soil and water Kd, organic carbon and water Koc, or octanol-water partition coefficient Kow) to elucidate processes in nature, but these are usually simple models that do not consider specificity of sorption sites or competition among molecules and elements (IAEA et al., 2009; Wu et al., 2010a).
The concentration and physicochemical properties of the contaminants will also determine the fate and transport in soils. As mentioned, a contaminant’s chemical nature (e.g. volatility, ionization, hydrophobicity, lipophilicity) will determine interaction with the soil matrix, and concentration will determine soil saturation; but size and spatial configuration will limit mobility through soil pores.
Soil contaminants can also be lost from the soil surface to the surrounding environment through wind and water erosion and from surface and subsurface soil via leaching to deeper soil depths. Contaminants can be dissolved in water from the soil surface and washed away, or can be transported in surface and shallow subsurface (tile) water entrained with mineral soil particles and dissolved organic matter to which they are adsorbed (Ciffroy, 2018). The erodibility of the soil surface therefore influences the surficial transport of soil contaminants, and soil management practices can strongly influence soil erosion risk. The following sections present the main factors influencing the transport of soil contaminants to adjacent and distance terrestrial and aquatic environments.
Table 4. The effects of selected soil properties on the mobilization of contaminants in soil
2.2.4. Soil pH
Soil pH regulates the surface charge of soil constituents and contaminant solubility. In acidic soils, clay surface edges, iron, manganese and aluminium (hydro-)oxides and amino (-NH2) groups in organic matter are protonated (that is, gain a proton H+), so an abundance of positive charges are available to attract anions on the binding sites. In contrast, alkaline soils present a dominant deprotonation of the OH- groups on variable charge mineral surfaces or on the phenolic (-OH) and carboxylic (-COOH) groups of organic matter, resulting in the attraction of cations to the exchange sites, with a greater adsorption the higher the pH (Alloway, 2013).
Trace element speciation, solubility and availability are mainly influenced by soil pH (Bourg and Loch, 1995). The form in soils will determine bioavailability and hence toxicity to plants, micro and macro-organisms, including humans (Adriano, 2001). Depending on the pH, metals and metalloids are present as free ions or complexed to a colloid in the soil solution, in gas phase, associated with the solid phases of the soil, or precipitated (Alloway, 2013). Thermodynamic processes that determine the bioavailability of trace elements are complex and will not be described here, but more information can be found in Adriano, 2001; Kabata-Pendias, 2010; Kabata-Pendias and Szteke, 2015; Roberts, Nachtegaal and Sparks, 2005.
Soil pH buffering capacity (pHBC) refers to the resilience to sudden changes in pH when an acid or alkaline solution is added to the soil. Calcium and magnesium, as well as sodium, potassium and ammonium, are the first cations to be released to neutralize the acidification effect, but when these cations are exhausted and the pH reduces, aluminium –and iron in extreme cases– start to be released from the oxides into the soil solution, causing toxicity (Bowman et al., 2008; Qiu, Wu and Zhang, 1998; Shindo and Fumoto, 1998). The dissolution of minerals and oxides by continued acid additions (as is the case in acid mine drainage) will lead to a permanent acidification and degradation of soil structure and soil functions (Nelson and Su, 2010). The soil acid buffer capacity is influenced by the soil CEC, the presence of carbonates and the exchangeable cations (Luo et al., 2015). The introduction of exogenous alkali (such as the addition of lime or gypsums to agricultural soils to raise pH) in soils will activate exchangeable anions and the active oxygen-containing functional groups, such as carboxyl, phenolic hydroxyl, and alcoholic hydroxyl, in the organic matter. Therefore the CEC/AEC and content and type of soil organic matter are the major factors responsible for alkali buffering capacity (Huang et al., 2009). The pHBC will therefore control the dissolution and speciation of contaminants, such as trace elements.
2.2.5. Soil organic matter (SOM)
Soil organic matter (SOM) comprises a wide range of organic molecules in different states of mineralization and complexation within the soil matrix, which will behave differently when interacting with contaminants. For example, dissolved organic matter forms complexes with contaminants, favouring movement with the soil solution, while humified organic matter (humic substances) can retain the contaminants, reducing availability and toxicity. Organic macromolecules that form soil organic matter (SOM) contain many pH- and redox-dependent functional groups, such as carboxylic acids (-COOH), alcohols and phenols (-OH), or amines (-NH2). These functional groups have a major role on the sorption of ionizable organic contaminants as well ionic forms of trace elements through covalent and hydrogen bonding, reducing accessibility to microbial interactions (Naidu and Bolan, 2008). Small organic compounds such as amino acids, sugar acids, short chain aliphatic acids and phenols have the capacity to form stable chelates with trace elements (McCauley, Jones and Jacobsen, 2009; Stevenson, 1972), and contaminants can also complex with aluminium and iron oxides (Pédrot et al., 2008).
The greater the concentration of organic matter, the greater the capacity of the soil to retain and immobilize contaminants, but this is also influenced by the complexity and polymerization (aromaticity) of SOM, with the most recalcitrant compounds (kerogens, biochar) showing the maximum adsorption capacity, and the geologically younger organic matter (plant residues or other biological residues) showing the least adsorption capacity (Pédrot et al., 2008; Weber, Huang and Yu, 1998). In soils with a low clay content, SOM plays a fundamental role in the immobilization of soil contaminants. The SOM also plays a role in the mobilization of contaminants through the soil profile but predominance of stable and humified organic matter over dissolved organic fractions contributes to the retention of contaminants in the topsoil layer and limits transfer to subsoil and groundwater (Okkenhaug et al., 2018; Pontoni et al., 2016).
In the case of non-ionic organic contaminants, soil pH and clay mineralogy have less of an influence. Sorption to SOM through covalent bond formation or through physical occlusion, depends on contaminant hydrophobicity and three-dimensional structure. Many studies have observed a positive linear relationship between the content of SOM and the sorption of hydrophobic organic contaminants (Ertli, Marton and Földényi, 2004; He et al., 2006; Karickhoff, Brown and Scott, 1979; Naidu and Bolan, 2008; Simpson, 2006; Wilson and Naidu, 2008).
Wastewater, manure and sewage sludge are sources of exogenous organic matter that impacts retention and bioavailability of soil contaminants, in addition to sourcing contaminants to soil (Bernal, Clemente and Walker, 2007; Börjesson and Kätterer, 2018). Although the accumulation and persistence of contaminants may be enhanced with the addition of active organic matter and the increase in cation exchange capacity (Placek, Grobelak and Kacprzak, 2016; Verlicchi and Zambello, 2015), exogenous SOM increases the C:N ratio and therefore promotes microbial activity to degrade organic contaminants (Włodarczyk-Makuła, 2010). Wołejko et al. (2018) observed 68 percent reduction of carcinogenic PAHs in urban soils after two-years of sewage application related to a more active microbial degradation.
Soil pH also regulates the solubility of SOM and therefore the concentration of any SOM-contaminant complexes. As mentioned by (Brümmer, 1986), SOM can show a higher potential to retain trace elements at very acidic conditions than clays and other soil minerals, but these will become mobile as the pH increases and the SOM solubilizes (Kalbitz and Wennrich, 1998). The increase in dissolved organic matter is also related to an increase in the mobility of hydrophobic organic contaminants such as PAHs (Kalbitz, Popp and Knappe, 1995). The stability of metallic complexes with soil fulvic acids changes with the pH (Schnitzer and Skinner, 1967), decreasing at pH 3.5 according to the following order: Cu2+ > Fe2+ > Ni2+ > Pb2+ > Co2+ > Ca2+ > Zn2+ > Mn2+ > Mg2+; whereas at pH 5.0 the order was: Cu2+ > Pb2+ > Fe2+ > Ni2+ > Mn2+ ≈ Co2+ > Ca2+ > Zn2+> Mg2+ (Smith, 1999).
2.2.6. Soil texture and mineralogy
Texture, and in particular clay type and content, importantly influences the sorption capacity of soils. Sand and silt have no or minimal cation and anion exchange capacity and influence primarily the soil structure and macroporosity. Sandy soils will permit greater and more rapid horizontal and vertical migration of contaminants within the soil profile, for both inorganic and organic contaminants (Krauss and Wilcke, 2002). On the other hand, clays are less permeable and possess ion exchange sites responsible for contaminant retention some of which are pH-dependent (Alamgir, 2016).
Clay minerals may be silicate or non-silicate. Silicate clays are formed by layers of silicates (phyllosilicates) organized in tetrahedral and octahedral sheets, coordinated by aluminium or silica cations, and held together by van der Waals bonds. Octahedral sheets are coordinated by cations; the most common are Ca, Mg, Al, Mg, and Fe (Kodama and Grim, 2014). Three main types of silicate clay minerals exist in the clay particle size fraction (<0.002 mm) (see Barton and Karathanasis (2002) and Osman (2013) for details) (Figure 17):
- 1:1 silicate clays: one tetrahedral and one octahedral sheets in which the anions at the exposed surface of the octahedral sheet are hydroxyls (e.g. kaolinite);
- 2:1 silicate clays: one octahedral sheet interspersed with two tetrahedral sheets (e.g. smectite, vermiculite, montmorillonite);
- 2:1:1 silicate clays: basic 2:1 layer structure with an interlayer sheet of brucite (Mg(OH)2) or gibbsite (Al(OH)3) (e.g. chlorite); and
Non-silicate clay minerals include Fe, Mn and Al (hydro-)oxides (e.g. hematite, gibbsite), calcite, sulphides (e.g. pyrite) and gypsum. (Hydro-)oxides and amorphous minerals show significant adsorption capacity for ionic contaminants (Alamgir, 2016).
The major difference among minerals in the clay size fraction, related to contaminant binding capacity, lies in the number and stability (permanent or variable) of binding sites (charged surfaces). For both ionic and organic contaminants, these sites are more prevalent in 2:1 and 2:1:1 clay types and allophanes (Stevenson, 1972). Permanently charged negative surfaces originate from isomorphic substitution of Al3+ for the Si4+ on the interlayer clay minerals, or from Fe2+/Mg2+ for the Al3+ in the octahedral layer. Cations and cationic contaminants bind to the negative charges. The charge on variable charge surfaces (e.g. silicate clay edges and surfaces of metal (hydro-)oxides) is determined by pH and the ionization of surface functional groups, being negative at high pH and positive at low pH. Additionally, in the case of trace elements, binding affinity varies not only with the type of clay and charged surfaces but also with the valence and atomic weight of the element, following the hierarchy of affinity Cu2+ > Cd2+ > Fe2+ > Pb2+ > Ni2+ > Co2+ > Mn2+ > Zn2+ (Fijałkowski et al., 2012).
Polar organic molecules can be retained at clay surfaces by hydrogen bonding with the hydration water on external surfaces (outer-sphere complex) or through electron donor-acceptor interactions (Sheng et al., 2001). Clay surfaces also show an affinity for neutral and non-polar organic molecules through hydrophobic interactions (Sheng et al., 2001 and reference herein). Clays may retain radionuclides more efficiently than SOM, and leaching is relatively easy (Fujii et al., 2014). It has been observed that the accumulation and stabilization of radionuclides over long periods in the soil depends on the predominant clay minerals, with 2:1 clays possessing additional sorption sites (Fujii et al., 2014; Giannakopoulou et al., 2012).
Figure 17. Main silicate clay structures.

2.2.7. Soil moisture
The water present in the soil matrix competes with air for pore space, and thereby reduces the availability of oxygen, which will affect soil organisms and the interactions with contaminants. Pore water also influences the redox conditions of the soil, which ultimately determines the oxidation state of contaminants and hence solubility and bioavailability (Husson, 2013; Masscheleyn, Delaune and Patrick, 1991). Soil redox potential can change with variations of saturation (naturally or intentionally such as flooding rice paddy soils), or due to changes in organic matter content and biological activity (Bourg and Loch, 1995). For example, paddy soils are subject to cycles of flooding and drying, which affects the redox potential but also pH, organic carbon content and contaminant speciation (Pan et al., 2016).
Transport of hydrophilic contaminants, such as the herbicide atrazine, within the soil profile occurs with the movement of soil water and dissolved ions, via matrix flow through available pore space as well as via preferential flow through cracks, earthworm burrows, root channels and other macropores (Flury, 1996; Jury and Nielsen, 1989). Hydrophobic contaminants such as PHCs can also be transported through preferential flow pathways directly at high concentrations or when adsorbed by or absorbed to dissolved and colloidal soil components (Gjettermann et al., 2009; Kögel-Knabner and Totsche, 1998; Li and Zhou, 2010; Roulier and Jarvis, 2003). Poorly soluble organic contaminants in water form a non-aqueous phase liquid, which tends to move downwards by gravity, displacing pore air and is potentially retained by capillary forces (Leharne, 2019).
The water content inside the soil pores and the soil permeability, determined by texture and structure, will strongly influence the risk and pathway of contaminant transport through the soil profile (Badin, Bedell and Delolme, 2009; Knappenberger et al., 2014). Sandy soils and soils with high macroporosity (including channels formed by roots, earthworms, and other macroorganisms and cracks formed by expansive clays), and with low organic matter content, will be more prone to leaching of contaminants, both soluble and hydrophobic, than those soils with a more developed pore structure, more reactive soil particles and higher organic carbon content, in which the processes of sorption to organo-mineral structures and the physical entrapment in small pores and aggregates predominate (Foster, Chilton and Stuart, 1991; Roulier and Jarvis, 2003). Similarly, transport of contaminants with water through the soil matrix and leaching deeper through the soil profile and to groundwater will be favoured in soils saturated or above field capacity, and will be much lower in drier soils close to or below the permanent wilting point (Badin, Bedell and Delolme, 2009; Knappenberger et al., 2014). The movement of contaminants up and down the soil profile is therefore strongly determined by soil moisture and inputs (rainfall, irrigation) and outputs (evapotranspiration, upward capillarity) of water in the soil system, as well as by diffusion (Ciffroy, 2018).
Water content strongly influences the pH and redox state, which both strongly affect solubility of contaminants. For example, lead, cadmium and zinc solubility is reduced at constant pH with an increment in redox potential, while under the same redox conditions, solubility is increased under acidic conditions (Husson, 2013). In general, most trace elements that form cations in solution show a higher solubility and therefore higher bioavailability under low pH and oxidized conditions. Arsenic, however, forms anionic complexes in solution and is more soluble and bioavailable at alkaline soil pH and under certain conditions of saturation when in the form of arsenite (As(III)). Mercury is highly soluble in its oxidized form (Hg2+), which can be complexed by SOM or absorbed by plants. Soil organic matter, as a major electron donor, will also influence the redox potential of the soil (Husson, 2013), influencing the fate and transport of contaminants (Stevenson, 1972).
2.2.8. Soil temperature
Soil temperature and its variation due to the influence of climate change can also have a significant impact on the fate and transport of soil contaminants (Noyes et al., 2009). There is evidence that a warmer global climate will increase the negative effects of soil pollution on ecosystems (Kozlov and Zvereva, 2011). Changes in temperature moderate the rate of water evapotranspiration from the soil surface, favouring the volatilization of some contaminants and affecting communities of soil organisms due to both the increase of temperature and the reduction of soil water content (Barmentlo et al., 2017). In addition, temperature is one of the main drivers of contaminant degradation (Ciffroy, 2018). Increases in temperature can contribute to accelerated microbial metabolism favouring the degradation of organic contaminants such as POPs and other pesticides (Noyes et al., 2009), but in contrast soil communities may also be stressed and degradation rates decreased (Velki and Ečimović, 2015). Although dynamics of SOM turnover in response to increased temperatures are predicted to be modified by the protective action of soil aggregates and organo-mineral complexes, but, under certain conditions, the acceleration of microbial metabolism with increasing temperature could be expected to accelerate the release of contaminants that were previously bound to SOM and influence other soil properties, such as the redox state or pH, causing a change in the mobility and availability of contaminants (Biswas et al., 2018).
2.2.9. Soil biodiversity
Soil organisms and plants influence contaminant fate and transport. Plants are the major source of biological litter and SOM, and can influence the mobilization of contaminants by rhizosphere changes in pH, redox and SOM characteristics. This changes contaminant adsorbed and accumulated in the below- and above-ground tissues. If plant material is not harvested, contaminant may be cycled to the soil as dead organic matter and incorporated into the topsoil organic layer (Wu et al., 2010b). Some plants have the capacity to stabilize organic and inorganic contaminants through root exudates and to filtrate and accumulate contaminants in the root zone or root tissues. Some contaminants may also be translocated to aerial tissues, stored or degraded in plant tissues, and volatilized through the stomata, e.g. mercury (Rodrigues et al., 2012). Figure 18 represents the different mechanisms used by plants to reduce the toxicity and effects of soil contaminants, mechanisms that are being leveraged to remediate polluted soil using bioremediation (see chapter 7).
Figure 18. Plant mechanisms for the uptake and stabilization of organic and inorganic contaminants.

Small mammals, insects, earthworms and other soil-dwelling macro-organisms contribute to the movement of contaminants by ingesting soil particles and SOM, and excreting into burrows and channels as they move through the soil profile (Goix et al., 2015; Leveque et al., 2014). Contaminant redistribution can be bidirectional: soil organisms can translocate contaminants from the soil surface into deeper layers, or to the surface from deeper layers (e.g. from geogenically contaminated deeper soil or soil affected by diffusion from contaminated groundwater) (Zorn, van Gestel and Eijsackers, 2008). In addition, soil organisms constitute the entry point of soil contaminants into the terrestrial food webs (Huerta-Lwanga et al., 2017; Sterckeman et al., 2000; Yung et al., 2019).
In degradation processes, microorganisms release organic compounds and also metabolic by-products that serve as chelating agents for trace elements, thereby influencing contaminant solubility. Microorganisms can utilize organic contaminants as a carbon source and substrate of enzymatic activities, thereby either degrading contaminants to their constituent elements or degrading them to less (or more) toxic metabolites. Microbial (bacterial and fungi) communities in the rhizosphere can also enhance organic contaminant uptake by plants. As with all organisms, from plants to bacteria, microorganisms excrete redox enzymes as part of their metabolism and mechanism of auto-defence. These enzymes may contribute, either, to the acidification of soil pH, increasing the mobility of some contaminants to facilitate adsorption, or to the reduction and precipitation of trace elements into less toxic forms (Boteva et al., 2016; Wu et al., 2010b).
- 1 A half-life is the time it takes for a certain amount of a contaminant to be reduced by half. This occurs as it dissipates or breaks down in the environment. When specifically referring to radionuclides, half-life indicates the amount of time it takes for the radionuclide to lose half of its radioactivity.
- 2 Volatility of a substance indicates how readily it converts into its gaseous form. At a given temperature and pressure, a substance with high volatility is more likely to exist as a gas, while a substance with low volatility is more likely to be a liquid or solid.
- 3 Lipophilicity is the key physicochemical parameter indicating the solubility of a substance into oils, fats and other non-polar substances such as lipids that make up biological membranes; solubility through the latter represents an important route for contaminant absorption and distribution in living organisms.
- 4 This report uses the term trace elements, which encompasses potentially toxic elements, heavy metals, persistent inorganic contaminants, and inorganic contaminants, all of which are defined in the Glossary.
- 5 The term “aliphatic” refers to any organic compound in which the carbon and hydrogen atoms are connected by single, double, or triple bonds to form non-ringed structures.
- 6 The term “aromatic” refers to any organic compound in which the carbon and hydrogen atoms are forming a planar unsaturated ring of atoms that is stabilized by an interaction of the bonds forming the ring.
- 7 This effect is caused by the influence of the volume of a functional group of a molecule in the course of a chemical reaction, in the conformation or in the intermolecular interactions of a molecule. The presence of a functional group can cause stereochemical impediment, repulsion and attraction, modifying the final conformation of the different isomers (McFarland and Clarke, 1989).
- 8 The Green Revolution refers to the dramatic advances in research and technology in agriculture, such as the development and use of genetically improved varieties, increased use of fertilizers and pesticides, improved irrigation, the use of heavy machinery that facilitated ploughing, sowing and harvesting, etc. that resulted in greatly increased crop yields (IFPRI, 2002).
- 9 Biomagnification is the accumulation of a contaminant by an organism from both environmental and dietary exposure, resulting in a higher concentration than would have been the case from exposure to the environment alone. When biomagnification is occurring, the concentration of a contaminant is higher in an organism than in organisms at lower levels of the food chain.